COM_STUDIES_DEFAULT_PAGE_TITLE
Nordrhein-Westfalen
Aachen
Universitätsklinikum Aachen, Medizinische Klinik IV, Hämatologie und Onkologie
02788-47851
Studien
AlloRelapseMM
Allogene Stammzelltransplantation im Vergleich zur konventionellen Therapie als Salvage-Therapie für Patienten mit rezidiviertem / progredientem Multiplen Myelom nach einer Erstlinientherapie (AlloRelapseMMStudy)
Organisatorische Daten:
| Prüfplancode: | |
| ISRCTN: | |
| EudraCT: | 2021-001--5-67 |
| Clinicaltrials.gov: | NCT0567319 |
| DRKS: | |
| Sponsor: | Universitätsklinikum Hamburg-Eppendorf mit Unterstützung des Gemeinsamen Bundesausschusses (G-BA) |
| Studienphase: | III |
| Status: | Rekrutierung läuft, geplant bis 6/2027 |
Ziel:
In dieser Studie wird untersucht, ob die Transplantation von gespendeten Stammzellen (allogene Stammzelltransplantation) Vorteile gegenüber der gängigen Therapie bei Patienten mit Multiplem Myelom bietet, bei denen es nach einer Transplantation von körpereigenen Stammzellen (autologe Stammzelltransplantation) als Erstlinientherapie zu einem Erkrankungsrückfall (Rezidiv) oder einem Fortschreiten der Krankheit (Progression) gekommen ist. Es wird geprüft, ob die allogene Stammzelltransplantation das Gesamtüberleben bei diesen Patienten verlängert.
Hintergrund:
Eine Behandlungsoption des Multiplen Myeloms ist die Stammzelltransplantation. Nach einer Chemotherapie, bei der die Krebszellen zerstört werden, werden körpereigene (autologe) oder gespendete körperfremde (allogene) Stammzellen übertragen. Die übertragenen Stammzellen siedeln sich dann im Knochenmark an und bilden „frische“ Blutzellen. Die Stammzelltransplantation kommt als Behandlungsoption nach der ersten Diagnose (Erstlinientherapie), aber auch bei einem Rückfall nach Vorbehandlung (jenseits der Erstlinientherapie) in Frage. Auch wenn das Multiple Myelom oft nicht endgültig heilbar ist, soll die Therapie die Lebenserwartung verlängern und die Symptome zurückdrängen. (Quelle: https://www.g-ba.de/studien/erprobung/allorelapsemmstudy-studie/)
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- Diagnose Multiples Myelom,
- Rückfall (Rezidiv) oder Fortschreiten (Progress) des Multiplen Myeloms unter oder nach einer autologen Stammzelltransplantation
- Erhalt von höchstens 1 Zyklus einer sogenannten Salvage-Therapie (Rezidiv-Therapie) vor Einschluss in die Studie
Ausschlusskriterien:
- Unzureichende Organfunktion gemäß der Definition im Prüfplan
- Aktive Hepatitis B oder C oder unkontrollierte HIV-Infektion
- Andere aktive bösartige Erkrankung
- Vorherige Behandlung mit allogenen Stammzellen
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Induktionstherapie mit Lenalidomid, Bortezomib und Dexamethason und entweder intravenösem oder subkutanem Isatuximab bei Patienten mit neu diagnostiziertem Multiplem Myelom, die für eine Hochdosis-Chemotherapie mit nachfolgender autologer Stammzelltransplantation geeignet sind
Organisatorische Daten:
| Prüfplancode: | GMMG-HD 8 / DSMM XIX |
| ISRCTN: | |
| EudraCT: | 2022-000996-38 |
| Clinicaltrials.gov: | NCT05804032 |
| DRKS: | |
| Sponsor: | Universitätsklinikum Heidelberg DSMM, Sanofi und KKS-Netzwerk |
| Studienphase: | |
| Status: | Rekrutierung geplant bis Juni 2025 |
Ziel:
In dieser Studie für Patienten mit zuvor unbehandeltem Multiplem Myelom soll die Wirksamkeit der Induktionstherapie mit Isatuximab in Kombination mit Lenalidomid/Bortezomib/Dexamethason (RVd) bei Gabe von Isatuximab unter die Haut (subkutan) gegenüber einer Gabe in die Vene (intravenös) untersucht werden.
Durchführung:
Randomisierung: Die Patienten werden vor der Induktionstherapie nach dem Zufallsverfahren (randomisiert) einem von 2 Studienarmen (A oder B) zugeordnet.
- Patienten in Arm A erhalten 3 Zyklen des monoklonalen Antikörpers Isatuximab intravenös in Kombination mit dem RVd-Schema (Lenalidomid, Bortezomib, Dexamethason). Jeder Zyklus hat eine Dauer von 42 Tagen.
- Patienten in Arm B erhalten 3 Zyklen Isatuximab subkutan in Kombination mit RVd.
Nach der Induktionstherapie erhalten die Patienten eine Standard-Intensivierung (in der Regel eine Mobilisierungstherapie auf Cyclophosphamid-Basis, Stammzellenentnahme und hochdosiertes Melphalan, gefolgt von einer autologen Stammzelltransplantation
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- Bestätigte Diagnose eines neu diagnostizierten behandlungsbedürftigen Multiplen Myeloms gemäß den überarbeiteten Diagnosekriterien der International Myeloma Working Group (IMWG))
- Der Patient ist für eine Hochdosis-Chemotherapie und eine autologe Stammzelltransplantation geeignet
- Messbare Krankheitsaktivität (gemäß der Definition im Prüfplan)
- Alter 18 bis 70 Jahre bei Studieneinschluss
Ausschlußkriterien:
- Bekannte Überempfindlichkeit (oder Kontraindikation) gegen einen der Bestandteile der Studientherapie
- Systemische Amyloid-Leichtketten-Amyloidose (außer lokalisierte Amyloid-Leichtketten -Amyloidose, die auf die Haut oder das Knochenmark beschränkt ist)
- Plasmazell-Leukämie
- Vorherige Chemo- oder Strahlentherapie in den letzten 5 Jahren (außer lokaler Strahlentherapie bei lokalem Fortschreiten des Myeloms)
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Studie zu Elranatamab als Monotherapie und Elranatamab + Daratumumab im Vergleich zu Daratumumab + Pomalidomid + Dexamethason bei Teilnehmern mit rezidiviertem/refraktärem Multiplem Myelom
Organisatorische Daten:
| Prüfplancode: | C1071005, MagnetisMM-5 |
| ISRCTN: | |
| EudraCT: | 2021-000044-22 |
| Clinicaltrials.gov: | NCT05020236 |
| DRKS: | |
| Sponsor: | Pfizer |
| Studienphase: | Phase 3 |
| Status: | Rekrutierung läuft, geplant bis Okt. 2024 |
Ziel:
Mit dieser Studie soll untersucht werden, ob der bispezifische BCMA/CD3-Antikörper Elranatamab allein und/oder in Kombination mit dem monoklonalen Anti-CD38-Antikörper Daratumumab für Patienten mit Multiplem Myelom einen größeren Nutzen bringt als eine Kombinationstherapie aus Daratumumab, Pomalidomid und Dexamethason.
Hintergrund:
Der bispezifische BCMA/CD3-Antikörper Elranatamab (PF-06863135) zeigte bei Patienten mit rezidiviertem/refraktärem Multiplem Myelom eine vielversprechende vorläufige Wirksamkeit und Verträglichkeit.
Daten aus der Phase-1-Studie MagnetisMM-1 (NCT03269136), die auf der ASCO-Jahrestagung 2021 vorgestellt wurden, zeigten, dass Elranatamab in Dosen von bis zu 1000 µg/kg ein überschaubares Sicherheitsprofil ohne dosislimitierende Toxizitäten aufweist.
Darüber hinaus erzielte der Wirkstoff bei Dosierungen von 215 µg/kg und höher eine Gesamtansprechrate (ORR) von 70 % und eine vollständige Ansprechrate (CR)/stringente CR von 30 %. Bei der empfohlenen Phase-2-Dosis von 1000 µg/kg erzielte der Wirkstoff eine ORR von 83,3 %. Bemerkenswert ist, dass drei von vier in die Studie aufgenommenen Patienten, die zuvor mit einer BCMA-gerichteten Therapie behandelt worden waren, ein Ansprechen erzielten.
Durchführung:
In die Studie werden Patienten mit Multiplem Myelom aufgenommen, die zuvor mit Lenalidomid und einem Proteasom-Inhibitor behandelt wurden.
In Teil 1 der Studie wird die Sicherheit und Wirksamkeit verschiedener Dosen von Elranatamab in Kombination mit Daratumumab untersucht.
Die Teilnehmer an Teil 2 der Studie werden nach dem Zufallsprinzip einer von drei Behandlungsgruppen zugeteilt.
- Teilnehmer in Gruppe 1 erhalten Elranatamab allein als Injektion unter die Haut.
- Teilnehmer der Gruppe 2 erhalten Elranatamab und Daratumumab als Injektion unter die Haut.
- Teilnehmer der Gruppe 3 erhalten die Standardtherapie Daratumumab als Injektion unter die Haut sowie Pomalidomid und Dexamethason zur Einnahme.
In Teil 2 wird die Sicherheit und Aktivität von (1) Elranatamab allein im Vergleich zu Daratumumab, Pomalidomid und Dexamethason und (2) Elranatamab plus Daratumumab im Vergleich zu Daratumumab, Pomalidomid und Dexamethason verglichen. Die Teilnehmer beider Studienteile erhalten die Studienbehandlung so lange, bis ihre Krankheit fortschreitet, unannehmbare Nebenwirkungen auftreten oder sie sich entscheiden, nicht weiter an der Studie teilzunehmen.
Einschlusskriterien:
- Diagnose eines Multiplen Myeloms gemäß den von der International Myeloma Working Group (IMWG) aufgestellten Diagnosekriterien )
- Messbare Erkrankung, definiert durch mindestens 1 der folgenden Punkte:
- M-Protein im Serum > 0,5 g/dl durch SPEP
- M-Protein-Ausscheidung im Urin > 200 mg/24 Stunden mittels UPEP
- Freie Immunglobulin-Leichtketten (FLC) im Serum ≥ 10 mg/dl (≥ 100 mg/l) UND abnormes Verhältnis der Immunglobuline Kappa-zu-Lambda-FLC-im Serum
- Vorherige Therapie gegen multiples Myelom, einschließlich Behandlung mit Lenalidomid und einem Proteasom-Inhibitor.
- ECOG Performance Status (Index zur Abstufung der Lebensqualität von Patienten mit Krebserkrankungen) von 0, 1 oder 2.
Ausschlusskriterien:
- Schwelendes Multiples Myelom
- Aktive Plasmazellleukämie
- Amyloidose
- POEMS-Syndrom
- Stammzelltransplantation innerhalb von 12 Wochen vor der Aufnahme in die Studie
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt
Bielefeld
Evangelisches Krankenhaus Bielefeld, Klinik für innere Medizin, Hämatologie /Onkologie und Palliativmedizin
0521-7727-5761
Studien
GMMG-HD8/DSMM-XIX
Induktionstherapie mit Lenalidomid, Bortezomib und Dexamethason und entweder intravenösem oder subkutanem Isatuximab bei Patienten mit neu diagnostiziertem Multiplem Myelom, die für eine Hochdosis-Chemotherapie mit nachfolgender autologer Stammzelltransplantation geeignet sind
Organisatorische Daten:
| Prüfplancode: | GMMG-HD 8 / DSMM XIX |
| ISRCTN: | |
| EudraCT: | 2022-000996-38 |
| Clinicaltrials.gov: | NCT05804032 |
| DRKS: | |
| Sponsor: | Universitätsklinikum Heidelberg DSMM, Sanofi und KKS-Netzwerk |
| Studienphase: | |
| Status: | Rekrutierung geplant bis Juni 2025 |
Ziel:
In dieser Studie für Patienten mit zuvor unbehandeltem Multiplem Myelom soll die Wirksamkeit der Induktionstherapie mit Isatuximab in Kombination mit Lenalidomid/Bortezomib/Dexamethason (RVd) bei Gabe von Isatuximab unter die Haut (subkutan) gegenüber einer Gabe in die Vene (intravenös) untersucht werden.
Durchführung:
Randomisierung: Die Patienten werden vor der Induktionstherapie nach dem Zufallsverfahren (randomisiert) einem von 2 Studienarmen (A oder B) zugeordnet.
- Patienten in Arm A erhalten 3 Zyklen des monoklonalen Antikörpers Isatuximab intravenös in Kombination mit dem RVd-Schema (Lenalidomid, Bortezomib, Dexamethason). Jeder Zyklus hat eine Dauer von 42 Tagen.
- Patienten in Arm B erhalten 3 Zyklen Isatuximab subkutan in Kombination mit RVd.
Nach der Induktionstherapie erhalten die Patienten eine Standard-Intensivierung (in der Regel eine Mobilisierungstherapie auf Cyclophosphamid-Basis, Stammzellenentnahme und hochdosiertes Melphalan, gefolgt von einer autologen Stammzelltransplantation
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- Bestätigte Diagnose eines neu diagnostizierten behandlungsbedürftigen Multiplen Myeloms gemäß den überarbeiteten Diagnosekriterien der International Myeloma Working Group (IMWG))
- Der Patient ist für eine Hochdosis-Chemotherapie und eine autologe Stammzelltransplantation geeignet
- Messbare Krankheitsaktivität (gemäß der Definition im Prüfplan)
- Alter 18 bis 70 Jahre bei Studieneinschluss
Ausschlußkriterien:
- Bekannte Überempfindlichkeit (oder Kontraindikation) gegen einen der Bestandteile der Studientherapie
- Systemische Amyloid-Leichtketten-Amyloidose (außer lokalisierte Amyloid-Leichtketten -Amyloidose, die auf die Haut oder das Knochenmark beschränkt ist)
- Plasmazell-Leukämie
- Vorherige Chemo- oder Strahlentherapie in den letzten 5 Jahren (außer lokaler Strahlentherapie bei lokalem Fortschreiten des Myeloms)
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Städtische Kliniken Bielefeld Mitte, Klinik für Hämatologie /Onkologie und Palliativmedizin
0521-581-0
Studiengesellschaft Onkologie Bielefeld GbR
Bochum
Medizinische Universitätsklinik Knappschaftskrankenhaus
0234-299-0
Onkologische Gemeinschaftspraxis Bückner /Nückel
0234-912810-0
Bonn
Johanniter-Krankenhaus Bonn
0228-5430
Universitätsklinikum Bonn, Med. Univ.-Klinik und Poliklinik III, Hämatologie und Onkologie
0228-287-17236
Studien
AlloRelapseMM
Allogene Stammzelltransplantation im Vergleich zur konventionellen Therapie als Salvage-Therapie für Patienten mit rezidiviertem / progredientem Multiplen Myelom nach einer Erstlinientherapie (AlloRelapseMMStudy)
Organisatorische Daten:
| Prüfplancode: | |
| ISRCTN: | |
| EudraCT: | 2021-001--5-67 |
| Clinicaltrials.gov: | NCT0567319 |
| DRKS: | |
| Sponsor: | Universitätsklinikum Hamburg-Eppendorf mit Unterstützung des Gemeinsamen Bundesausschusses (G-BA) |
| Studienphase: | III |
| Status: | Rekrutierung läuft, geplant bis 6/2027 |
Ziel:
In dieser Studie wird untersucht, ob die Transplantation von gespendeten Stammzellen (allogene Stammzelltransplantation) Vorteile gegenüber der gängigen Therapie bei Patienten mit Multiplem Myelom bietet, bei denen es nach einer Transplantation von körpereigenen Stammzellen (autologe Stammzelltransplantation) als Erstlinientherapie zu einem Erkrankungsrückfall (Rezidiv) oder einem Fortschreiten der Krankheit (Progression) gekommen ist. Es wird geprüft, ob die allogene Stammzelltransplantation das Gesamtüberleben bei diesen Patienten verlängert.
Hintergrund:
Eine Behandlungsoption des Multiplen Myeloms ist die Stammzelltransplantation. Nach einer Chemotherapie, bei der die Krebszellen zerstört werden, werden körpereigene (autologe) oder gespendete körperfremde (allogene) Stammzellen übertragen. Die übertragenen Stammzellen siedeln sich dann im Knochenmark an und bilden „frische“ Blutzellen. Die Stammzelltransplantation kommt als Behandlungsoption nach der ersten Diagnose (Erstlinientherapie), aber auch bei einem Rückfall nach Vorbehandlung (jenseits der Erstlinientherapie) in Frage. Auch wenn das Multiple Myelom oft nicht endgültig heilbar ist, soll die Therapie die Lebenserwartung verlängern und die Symptome zurückdrängen. (Quelle: https://www.g-ba.de/studien/erprobung/allorelapsemmstudy-studie/)
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- Diagnose Multiples Myelom,
- Rückfall (Rezidiv) oder Fortschreiten (Progress) des Multiplen Myeloms unter oder nach einer autologen Stammzelltransplantation
- Erhalt von höchstens 1 Zyklus einer sogenannten Salvage-Therapie (Rezidiv-Therapie) vor Einschluss in die Studie
Ausschlusskriterien:
- Unzureichende Organfunktion gemäß der Definition im Prüfplan
- Aktive Hepatitis B oder C oder unkontrollierte HIV-Infektion
- Andere aktive bösartige Erkrankung
- Vorherige Behandlung mit allogenen Stammzellen
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Eine zweistufige, randomisierte, multizentrische, offene Phase-3-Studie zum Vergleich von Mezigdomid (CC-92480), Bortezomib und Dexamethason (MEZIVd) mit Pomalidomid, Bortezomib und Dexamethason (PVd) bei Patienten mit rezidiviertem oder refraktärem Multiplem Myelom (RRMM): SUCCESSOR-1
Organisatorische Daten:
| Prüfplancode: | CA- 057-001 |
| ISRCTN: | |
| EudraCT: | 2021-001957-30 |
| Clinicaltrials.gov: | NCT05519085 |
| DRKS: | |
| Sponsor: | Bristol Meyer Squibb |
| Studienphase: | Phase 3 |
| Status: | Rekrutierung läuft, geplant bis Nov. 2025 |
Ziele:
Ziel der Studie ist der Vergleich der Wirksamkeit und Sicherheit von Mezigdomid (CC-92480), Bortezomib und Dexamethason (MeziVd) gegenüberPomalidomid in Kombination mit Bortezomib und Dexamethason (PVd) bei Teilnehmern mit rezidiviertem oder refraktärem Multiplem Myelom a (RRMM), die zuvor zwischen 1 bis 3 Therapielinien und bereits Lenalidomid erhalten haben.
Hintergrund:
CC-92480 ist ein sogenannter CRBN-Modulator [Cereblon-(CRBN-)E3-Ligase-Modulator (CELMoD)]. Dieser neuartige Modulator hat vielfältige Wirkungen und wirkt unter anderem stark immunmodulierend. Der Wirkstoff führt zu einem schnellen, tiefen und anhaltenden Zerfall von Ikaros und Aiolos – zwei Faktoren, die zum Überleben der Myelomzellen beitragen.
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- Der Teilnehmer hat eine dokumentierte Diagnose eines Multiplen Myeloms und eine messbare Erkrankung, gemäß der Definition im Protokoll
- Der Teilnehmer hat bereits 1 bis 3 vorherige Linien einer Myelom-Therapielinie erhalten.
- Der Teilnehmer hat ein Lenalidomid-haltiges Therapieschema erhalten.
- Der Teilnehmer hat auf mindestens eine vorherige Myelom-Therapie minimal oder besser angesprochen..
Ausschlusskriterien:
- Der Teilnehmer hat während der Behandlung oder innerhalb von 60 Tagen nach der letzten Dosis eines Proteasom-Inhibitors einen Krankheitsfortschritt (Progression) erlitten. Dies gilt nicht für Teilnehmer, die während der Behandlung mit oder innerhalb von 60 Tagen nach der letzten Dosis einer alle 2 Wochen oder seltener verabreichten Bortezomib-Erhaltungstherapie eine Progression erlitten haben, sind nicht ausgeschlossen.
- Bei Teilnehmern, die zuvor mit einem Bortezomib-haltigen Schema behandelt wurden, war das beste erreichte Ansprechen kein minimales Ansprechen (MR) oder besser, oder der Teilnehmer setzte Bortezomib aufgrund von Toxizität ab.
- Der Teilnehmer wurde zuvor mit Mezigdomid oder Pomalidomid behandelt.
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt
Dortmund
St. Johannes-Hospital
0231-1843-0
Studien
DSMM XIVPhase III-Studie zum Vergleich einer Induktionstherapie aus Lenalidomid, Adriamycin, Dexamethason (RAD) mit Lenalidomid, Bortezomib, Dexamethason (VRD) gefolgt von einer am Ansprechen ausgerichteten Stammzelltransplantation bei Patienten mit neu diagnostiziertem multiplen Myelom.
| Prüfplancode: | DSMM XIV |
| ISRCTN: | |
| EudraCT: | 2009-016616-21 |
| Clincaltrials.gov: | NCT01685814 |
| DRKS: | 0003967 |
Allgemeine Informationen
In der Studie werden die Raten der kompletten Remission zweier Induktionsschemata (ein neues Medikament [RAD] gegen zwei neue Medikamente [VRD]) bei Patienten mit neu diagnostiziertem multiplem Myelom verglichen und das progressionsfreie Überleben nach einer Konsolidierungsbehandlung bestimmt.
Die Patienten sollten Über 18 Jahre und unter 65 Jahre alt sein, bisher ohne eine systemische Myelomtherapie sein und für eine Stammzelltransplantation in Frage kommen.
Die weiteren Ein- und Ausschlusskriterien kann man hier nachlesen.
Induktionstherapie mit Lenalidomid, Bortezomib und Dexamethason und entweder intravenösem oder subkutanem Isatuximab bei Patienten mit neu diagnostiziertem Multiplem Myelom, die für eine Hochdosis-Chemotherapie mit nachfolgender autologer Stammzelltransplantation geeignet sind
Organisatorische Daten:
| Prüfplancode: | GMMG-HD 8 / DSMM XIX |
| ISRCTN: | |
| EudraCT: | 2022-000996-38 |
| Clinicaltrials.gov: | NCT05804032 |
| DRKS: | |
| Sponsor: | Universitätsklinikum Heidelberg DSMM, Sanofi und KKS-Netzwerk |
| Studienphase: | |
| Status: | Rekrutierung geplant bis Juni 2025 |
Ziel:
In dieser Studie für Patienten mit zuvor unbehandeltem Multiplem Myelom soll die Wirksamkeit der Induktionstherapie mit Isatuximab in Kombination mit Lenalidomid/Bortezomib/Dexamethason (RVd) bei Gabe von Isatuximab unter die Haut (subkutan) gegenüber einer Gabe in die Vene (intravenös) untersucht werden.
Durchführung:
Randomisierung: Die Patienten werden vor der Induktionstherapie nach dem Zufallsverfahren (randomisiert) einem von 2 Studienarmen (A oder B) zugeordnet.
- Patienten in Arm A erhalten 3 Zyklen des monoklonalen Antikörpers Isatuximab intravenös in Kombination mit dem RVd-Schema (Lenalidomid, Bortezomib, Dexamethason). Jeder Zyklus hat eine Dauer von 42 Tagen.
- Patienten in Arm B erhalten 3 Zyklen Isatuximab subkutan in Kombination mit RVd.
Nach der Induktionstherapie erhalten die Patienten eine Standard-Intensivierung (in der Regel eine Mobilisierungstherapie auf Cyclophosphamid-Basis, Stammzellenentnahme und hochdosiertes Melphalan, gefolgt von einer autologen Stammzelltransplantation
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- Bestätigte Diagnose eines neu diagnostizierten behandlungsbedürftigen Multiplen Myeloms gemäß den überarbeiteten Diagnosekriterien der International Myeloma Working Group (IMWG))
- Der Patient ist für eine Hochdosis-Chemotherapie und eine autologe Stammzelltransplantation geeignet
- Messbare Krankheitsaktivität (gemäß der Definition im Prüfplan)
- Alter 18 bis 70 Jahre bei Studieneinschluss
Ausschlußkriterien:
- Bekannte Überempfindlichkeit (oder Kontraindikation) gegen einen der Bestandteile der Studientherapie
- Systemische Amyloid-Leichtketten-Amyloidose (außer lokalisierte Amyloid-Leichtketten -Amyloidose, die auf die Haut oder das Knochenmark beschränkt ist)
- Plasmazell-Leukämie
- Vorherige Chemo- oder Strahlentherapie in den letzten 5 Jahren (außer lokaler Strahlentherapie bei lokalem Fortschreiten des Myeloms)
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Duisburg
Bethesda-Krankenhaus, Standort BETHESDA
MVZ Duisberg
0203-7139 740
St. Johannes Hospital Duisburg, Medizinische Klinik II
Dr. A. Giagounidis, 0203-546-2442
Studien
GMMG-HD8/DSMM-XIX
Induktionstherapie mit Lenalidomid, Bortezomib und Dexamethason und entweder intravenösem oder subkutanem Isatuximab bei Patienten mit neu diagnostiziertem Multiplem Myelom, die für eine Hochdosis-Chemotherapie mit nachfolgender autologer Stammzelltransplantation geeignet sind
Organisatorische Daten:
| Prüfplancode: | GMMG-HD 8 / DSMM XIX |
| ISRCTN: | |
| EudraCT: | 2022-000996-38 |
| Clinicaltrials.gov: | NCT05804032 |
| DRKS: | |
| Sponsor: | Universitätsklinikum Heidelberg DSMM, Sanofi und KKS-Netzwerk |
| Studienphase: | |
| Status: | Rekrutierung geplant bis Juni 2025 |
Ziel:
In dieser Studie für Patienten mit zuvor unbehandeltem Multiplem Myelom soll die Wirksamkeit der Induktionstherapie mit Isatuximab in Kombination mit Lenalidomid/Bortezomib/Dexamethason (RVd) bei Gabe von Isatuximab unter die Haut (subkutan) gegenüber einer Gabe in die Vene (intravenös) untersucht werden.
Durchführung:
Randomisierung: Die Patienten werden vor der Induktionstherapie nach dem Zufallsverfahren (randomisiert) einem von 2 Studienarmen (A oder B) zugeordnet.
- Patienten in Arm A erhalten 3 Zyklen des monoklonalen Antikörpers Isatuximab intravenös in Kombination mit dem RVd-Schema (Lenalidomid, Bortezomib, Dexamethason). Jeder Zyklus hat eine Dauer von 42 Tagen.
- Patienten in Arm B erhalten 3 Zyklen Isatuximab subkutan in Kombination mit RVd.
Nach der Induktionstherapie erhalten die Patienten eine Standard-Intensivierung (in der Regel eine Mobilisierungstherapie auf Cyclophosphamid-Basis, Stammzellenentnahme und hochdosiertes Melphalan, gefolgt von einer autologen Stammzelltransplantation
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- Bestätigte Diagnose eines neu diagnostizierten behandlungsbedürftigen Multiplen Myeloms gemäß den überarbeiteten Diagnosekriterien der International Myeloma Working Group (IMWG))
- Der Patient ist für eine Hochdosis-Chemotherapie und eine autologe Stammzelltransplantation geeignet
- Messbare Krankheitsaktivität (gemäß der Definition im Prüfplan)
- Alter 18 bis 70 Jahre bei Studieneinschluss
Ausschlußkriterien:
- Bekannte Überempfindlichkeit (oder Kontraindikation) gegen einen der Bestandteile der Studientherapie
- Systemische Amyloid-Leichtketten-Amyloidose (außer lokalisierte Amyloid-Leichtketten -Amyloidose, die auf die Haut oder das Knochenmark beschränkt ist)
- Plasmazell-Leukämie
- Vorherige Chemo- oder Strahlentherapie in den letzten 5 Jahren (außer lokaler Strahlentherapie bei lokalem Fortschreiten des Myeloms)
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Zusätzliche Information
Institut für spezielle Pharmakotherapie
Düsseldorf
Klinik für Hämatologie und klinische Immunologie, UKD der Heinrich Heine Universität Düsseldor
0211- 81-17720
Studien
CC-220Studie mit Iberdomid
Eine Studie zur Bestimmung der Dosis und Verträglichkeit der CC-220 Monotherapie mit Iberdomid, in Kombination mit Dexamethason und in Kombination mit Dexamethason und Daratumumab oder Bortezomib bei Patienten mit fortgeschrittenem und refraktärem Multiplem Myelom (RRMM)
Organisatorische Daten:
| Prüfplancode: | CC-220-MM-001 |
| ISRCTN: | |
| EudraCT: | 2016-000860-40 |
| Clinicaltrials.gov: | NCT02773030 |
| DRKS: | |
| Sponsor: | Celgene |
| Studienphase: | Phase 1b/2a |
| Status: | Rekrutierung läuft, geplant bis Mai 2026 |
Ziel:
Dies ist eine multizentrische, länderübergreifende, offene Phase-1b/2a-Dosiseskalationsstudie zur Bestimmung der höchsten verträglichen Dosis, der Sicherheit, Verträglichkeit, Pharmakokinetik (Einwirkung des Körpers auf die Substanz)und Wirksamkeit von CC-220 (Iberdomid) als Monotherapie und in Kombination mit anderen Therapien bei Patienten mit rezidivierendem oder therapierefraktärem Multiplem Myelom.
Hintergrund:
Iberdomid (CC-220) ist eine neuartige Substanz, die in dieser Phase I/II Studie zur Behandlung von fortgeschrittenem und refraktärem Multiplem Myelom in Kombination mit anderen Therapien untersucht wird. Präklinische Studien zu Iberdomid zeigen, dass es wirksamer an das Protein Cereblon bindet als andere Cereblon-bindende Substanzen. Klinische Studien zu Bortezomib und Daratumumab in Kombination mit anderen Cereblon-bindenden Mitteln haben eine hohe Verträglichkeit bei bemerkenswerter Wirksamkeit bei RRMM gezeigt. Allerdings wurden diese Kombinationen mit Iberdomid bisher nicht untersucht. Insgesamt gesehen untermauern die vorliegenden präklinischen Daten die Untersuchung von Iberdomid in Kombination mit sowohl Bortezomib/Dexamethason als auch Daratumumab in klinischen Studien.
(Michael Amatangelo et al, Blood 2018. 132, 1935; http://www.bloodjournal.org/content/132/Suppl_1/1935?sso-checked=true
Durchführung:
Die Studie wird in zwei Teilen durchgeführt: Teil 1 mit Kohorte A und B und Teil 2 mit Kohorte C, D, E, F, G1 und G2.
In Teil 1 (Phase 1b) wird die maximal verträgliche Dosis von oralem CC-220 als Monotherapie (Kohorte A) und die von oralem CC-220 in Kombination mit oralem Dexamethason (Kohorte B) bestimmt. Außerdem wird in diesem Teil die empfohlene CC-220-Dosis für Teil 2 (Phase 2) ermittelt.
In Teil 2 werden folgende Therapien untersucht:
Kohorte C: CC-220 als Monotherapie in der empfohlenen Dosis für Teil 2
Kohorte D: CC-220 in der empfohlenen Dosis für Teil 2 in Kombination mit oralem Dexamethason
Kohorte E:CC-220 in Kombination mit oralem Dexamethason und intravenösem Daratumumab
Kohorte F: CC-220 in Kombination mit oralem Dexamethason und subkutanem Bortezomib
Kohorte G1: CC-220 in Kombination mit einmal wöchentlich verabreichtem intravenösem Carfilzomib und oralem Dexamethason
Kohorte G2: CC-220 in Kombination mit zweimal wöchentlich verabreichtem intravenösem Carfilzomib und oralem Dexamethason.
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- Dokumentierte Diagnose eines Multiplen Myeloms (MM) und messbare Krankheit, definiert als
- M-Protein: Serum-Elektrophorese ≥ 0,5 g/dl und/oder Urinprotein-Elektrophorese ≥ 200 mg/24 Stunden und/oder
- Leichtketten-Myelom ohne messbare Erkrankung im Serum oder Urin - Dokumentierte Krankheitsprogression an oder innerhalb von 60 Tagen seit der letzten Dosis der letzten Myelombehandlung
- ECOG (Index zur Abstufung der Lebensqualität von Patienten mit Krebserkrankungen) von 0, 1 oder 2
Ausschlußkriterien:
- Nicht ekretorisches oder oligosekretorisches Multiples Myelom
- Plasmazellenleukämie oder Amyloidose
- Bestimmte Laborwerte, die nicht im Normalbereich liegen
- Periphere Neuropathie ≥ Grad 2
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Belantamab-Mafodotin
Bewertung der Wirksamkeit und Sicherheit von Belantamab-Mafodotin, Bortezomib und Dexamethason gegenüber Daratumumab, Bortezomib und Dexamethason bei Teilnehmern mit rezidiviertem/refraktärem Multiplen Myelom
Organisatorische Daten:
| Prüfplancode: | 207503 |
| ISRCTN: | |
| EudraCT: | 2018-003993-29 |
| Clinicaltrials.gov: | NCT04246047 |
| DRKS: | |
| Sponsor: | GlaxoSmithKline |
| Studienphase: | III |
| Status: | Rekrutierung geplant bis Juni/2026 |
Kurzbeschreibung:
Hierbei handelt es sich um eine randomisierte, offene Phase-3-Studie zur Untersuchung der Sicherheit und Wirksamkeit von Belantamab-Mafodotin in Kombination mit Bortezomib/Dexamethason (Arm A) gegenüber Daratumumab in Kombination mit Bortezomib/Dexamethason (Arm B) bei Teilnehmern mit rezidivierendem multiplem Myelom..
Hintergrund:
Die amerikanische Arzneimittelbehörde FDA hat Belantamab-Mafodotin-blmf (GSK2857916; Blenrep) für die Behandlung von Patienten mit rezidiviertem oder refraktärem Multiplen Myelom zugelassen, die zuvor mit mindestens 4 Therapien behandelt wurden, darunter ein immunmodulatorisches Mittel, ein Proteasom-Inhibitor und ein Anti-CD38-Antikörper. Dies ist weltweit die erste Anti-BCMA-Behandlung, die bei dieser Patientenpopulation verfügbar ist.
Belantamab-Mafodotin ist gegen das B-Zell-Reifungsantigen (BCMA) gerichtet, das eine wichtige Rolle für das Überleben von Plasmazellen spielt und auf Zellen des Multiplen Myeloms exprimiert wird. Das Antikörper-Wirkstoff-Konjugat Belantamab-Mafodotin bindet an BCMA auf Myelom-Zelloberflächen, wodurch der Zellzyklus angehalten und eine antikörperabhängige zelluläre Zytotoxizität induziert wird.
Normalerweise fördert BCMA das Überleben von Plasmazellen durch Signalübertragung von zwei bekannten Liganden. Dieser Signalweg hat sich als wichtig für das Wachstum und Überleben von Myelomzellen erwiesen.
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- Bestätigte Diagnose eines Multiplen Myeloms (MM) gemäß IMWG-Kriterien
- Vorbehandlung des MM mit mindestens einer Therapielinie und dokumentierte Krankheitsprogression während oder nach der letzten Therapie.
- ECOG Performance Status (Index zur Abstufung der Lebensqualität von Patienten mit Krebserkrankungen) von 0 bis 2
- Muss mindestens 1 Aspekt einer messbaren Erkrankung aufweisen, wie folgt definiert:
- Urin-M-Protein-Ausscheidung > 200 mg pro 24 Stunden, oder
- Serum-M-Protein-Konzentration > 0,5 g/dl, oder
- Freie Leichtketten im Serum (FLC): beteiligter FLC-Spiegel > 10 mg/dl (> 100 mg/L) und ein abnormes Verhältnis der freien Leichtketten im Serum (< 0,26 oder > 1,65).
- Alle Nebenwirkungen, die infolge durch Vorbehandlungen hervorgerufen wurden, müssen zum Zeitpunkt der Rekrutierung < Grad 1 sein, mit Ausnahme von Alopezie.
Ausschlußkriterien:
- Unverträglichkeit gegenüber Daratumumab.
- Kein bzw. kein hinreichendes Ansprechen (therapierefraktär) gegenüber Daratumumab oder einer anderen Anti-CD38-Therapie (definiert als fortschreitende Erkrankung während der Behandlung mit einer Anti-CD38-Therapie oder innerhalb von 60 Tagen nach Abschluss dieser Behandlung).
- Unverträglichkeit gegenüber Bortezomib oder kein bzw. kein hinreichendes Ansprechen (therapierefraktär) gegenüber Bortezomib (definiert als fortschreitende Erkrankung während der Behandlung mit einem Bortezomib-haltigen Schema von 1,3 mg/m2 zweimal wöchentlich oder innerhalb von 60 Tagen nach Abschluss dieser Behandlung). Hinweis: Patienten mit fortschreitender Erkrankung während der Behandlung mit einem wöchentlichen Bortezomib-Schema dürfen teilnehmen.
- Anhaltende periphere Neuropathie von Grad 2 oder höher oder neuropathische Schmerzen.
- Vorherige Behandlung mit einer Anti-B-Zellreifungsantigen- (Anti-BCMA-) Therapie.
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Induktionstherapie mit Lenalidomid, Bortezomib und Dexamethason und entweder intravenösem oder subkutanem Isatuximab bei Patienten mit neu diagnostiziertem Multiplem Myelom, die für eine Hochdosis-Chemotherapie mit nachfolgender autologer Stammzelltransplantation geeignet sind
Organisatorische Daten:
| Prüfplancode: | GMMG-HD 8 / DSMM XIX |
| ISRCTN: | |
| EudraCT: | 2022-000996-38 |
| Clinicaltrials.gov: | NCT05804032 |
| DRKS: | |
| Sponsor: | Universitätsklinikum Heidelberg DSMM, Sanofi und KKS-Netzwerk |
| Studienphase: | |
| Status: | Rekrutierung geplant bis Juni 2025 |
Ziel:
In dieser Studie für Patienten mit zuvor unbehandeltem Multiplem Myelom soll die Wirksamkeit der Induktionstherapie mit Isatuximab in Kombination mit Lenalidomid/Bortezomib/Dexamethason (RVd) bei Gabe von Isatuximab unter die Haut (subkutan) gegenüber einer Gabe in die Vene (intravenös) untersucht werden.
Durchführung:
Randomisierung: Die Patienten werden vor der Induktionstherapie nach dem Zufallsverfahren (randomisiert) einem von 2 Studienarmen (A oder B) zugeordnet.
- Patienten in Arm A erhalten 3 Zyklen des monoklonalen Antikörpers Isatuximab intravenös in Kombination mit dem RVd-Schema (Lenalidomid, Bortezomib, Dexamethason). Jeder Zyklus hat eine Dauer von 42 Tagen.
- Patienten in Arm B erhalten 3 Zyklen Isatuximab subkutan in Kombination mit RVd.
Nach der Induktionstherapie erhalten die Patienten eine Standard-Intensivierung (in der Regel eine Mobilisierungstherapie auf Cyclophosphamid-Basis, Stammzellenentnahme und hochdosiertes Melphalan, gefolgt von einer autologen Stammzelltransplantation
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- Bestätigte Diagnose eines neu diagnostizierten behandlungsbedürftigen Multiplen Myeloms gemäß den überarbeiteten Diagnosekriterien der International Myeloma Working Group (IMWG))
- Der Patient ist für eine Hochdosis-Chemotherapie und eine autologe Stammzelltransplantation geeignet
- Messbare Krankheitsaktivität (gemäß der Definition im Prüfplan)
- Alter 18 bis 70 Jahre bei Studieneinschluss
Ausschlußkriterien:
- Bekannte Überempfindlichkeit (oder Kontraindikation) gegen einen der Bestandteile der Studientherapie
- Systemische Amyloid-Leichtketten-Amyloidose (außer lokalisierte Amyloid-Leichtketten -Amyloidose, die auf die Haut oder das Knochenmark beschränkt ist)
- Plasmazell-Leukämie
- Vorherige Chemo- oder Strahlentherapie in den letzten 5 Jahren (außer lokaler Strahlentherapie bei lokalem Fortschreiten des Myeloms)
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Zusätzliche Information
Marienhospital Düsseldorf
0211-4400-0
Studien
GMMG-HD8/DSMM-XIX
Induktionstherapie mit Lenalidomid, Bortezomib und Dexamethason und entweder intravenösem oder subkutanem Isatuximab bei Patienten mit neu diagnostiziertem Multiplem Myelom, die für eine Hochdosis-Chemotherapie mit nachfolgender autologer Stammzelltransplantation geeignet sind
Organisatorische Daten:
| Prüfplancode: | GMMG-HD 8 / DSMM XIX |
| ISRCTN: | |
| EudraCT: | 2022-000996-38 |
| Clinicaltrials.gov: | NCT05804032 |
| DRKS: | |
| Sponsor: | Universitätsklinikum Heidelberg DSMM, Sanofi und KKS-Netzwerk |
| Studienphase: | |
| Status: | Rekrutierung geplant bis Juni 2025 |
Ziel:
In dieser Studie für Patienten mit zuvor unbehandeltem Multiplem Myelom soll die Wirksamkeit der Induktionstherapie mit Isatuximab in Kombination mit Lenalidomid/Bortezomib/Dexamethason (RVd) bei Gabe von Isatuximab unter die Haut (subkutan) gegenüber einer Gabe in die Vene (intravenös) untersucht werden.
Durchführung:
Randomisierung: Die Patienten werden vor der Induktionstherapie nach dem Zufallsverfahren (randomisiert) einem von 2 Studienarmen (A oder B) zugeordnet.
- Patienten in Arm A erhalten 3 Zyklen des monoklonalen Antikörpers Isatuximab intravenös in Kombination mit dem RVd-Schema (Lenalidomid, Bortezomib, Dexamethason). Jeder Zyklus hat eine Dauer von 42 Tagen.
- Patienten in Arm B erhalten 3 Zyklen Isatuximab subkutan in Kombination mit RVd.
Nach der Induktionstherapie erhalten die Patienten eine Standard-Intensivierung (in der Regel eine Mobilisierungstherapie auf Cyclophosphamid-Basis, Stammzellenentnahme und hochdosiertes Melphalan, gefolgt von einer autologen Stammzelltransplantation
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- Bestätigte Diagnose eines neu diagnostizierten behandlungsbedürftigen Multiplen Myeloms gemäß den überarbeiteten Diagnosekriterien der International Myeloma Working Group (IMWG))
- Der Patient ist für eine Hochdosis-Chemotherapie und eine autologe Stammzelltransplantation geeignet
- Messbare Krankheitsaktivität (gemäß der Definition im Prüfplan)
- Alter 18 bis 70 Jahre bei Studieneinschluss
Ausschlußkriterien:
- Bekannte Überempfindlichkeit (oder Kontraindikation) gegen einen der Bestandteile der Studientherapie
- Systemische Amyloid-Leichtketten-Amyloidose (außer lokalisierte Amyloid-Leichtketten -Amyloidose, die auf die Haut oder das Knochenmark beschränkt ist)
- Plasmazell-Leukämie
- Vorherige Chemo- oder Strahlentherapie in den letzten 5 Jahren (außer lokaler Strahlentherapie bei lokalem Fortschreiten des Myeloms)
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Eschweiler
St. Antonius-Hospital Eschweiler, Akademisches Lehrkrankenhaus der RWTH Aachen
02403-76-0
Studien
GMMG-HD8/DSMM-XIX
Induktionstherapie mit Lenalidomid, Bortezomib und Dexamethason und entweder intravenösem oder subkutanem Isatuximab bei Patienten mit neu diagnostiziertem Multiplem Myelom, die für eine Hochdosis-Chemotherapie mit nachfolgender autologer Stammzelltransplantation geeignet sind
Organisatorische Daten:
| Prüfplancode: | GMMG-HD 8 / DSMM XIX |
| ISRCTN: | |
| EudraCT: | 2022-000996-38 |
| Clinicaltrials.gov: | NCT05804032 |
| DRKS: | |
| Sponsor: | Universitätsklinikum Heidelberg DSMM, Sanofi und KKS-Netzwerk |
| Studienphase: | |
| Status: | Rekrutierung geplant bis Juni 2025 |
Ziel:
In dieser Studie für Patienten mit zuvor unbehandeltem Multiplem Myelom soll die Wirksamkeit der Induktionstherapie mit Isatuximab in Kombination mit Lenalidomid/Bortezomib/Dexamethason (RVd) bei Gabe von Isatuximab unter die Haut (subkutan) gegenüber einer Gabe in die Vene (intravenös) untersucht werden.
Durchführung:
Randomisierung: Die Patienten werden vor der Induktionstherapie nach dem Zufallsverfahren (randomisiert) einem von 2 Studienarmen (A oder B) zugeordnet.
- Patienten in Arm A erhalten 3 Zyklen des monoklonalen Antikörpers Isatuximab intravenös in Kombination mit dem RVd-Schema (Lenalidomid, Bortezomib, Dexamethason). Jeder Zyklus hat eine Dauer von 42 Tagen.
- Patienten in Arm B erhalten 3 Zyklen Isatuximab subkutan in Kombination mit RVd.
Nach der Induktionstherapie erhalten die Patienten eine Standard-Intensivierung (in der Regel eine Mobilisierungstherapie auf Cyclophosphamid-Basis, Stammzellenentnahme und hochdosiertes Melphalan, gefolgt von einer autologen Stammzelltransplantation
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- Bestätigte Diagnose eines neu diagnostizierten behandlungsbedürftigen Multiplen Myeloms gemäß den überarbeiteten Diagnosekriterien der International Myeloma Working Group (IMWG))
- Der Patient ist für eine Hochdosis-Chemotherapie und eine autologe Stammzelltransplantation geeignet
- Messbare Krankheitsaktivität (gemäß der Definition im Prüfplan)
- Alter 18 bis 70 Jahre bei Studieneinschluss
Ausschlußkriterien:
- Bekannte Überempfindlichkeit (oder Kontraindikation) gegen einen der Bestandteile der Studientherapie
- Systemische Amyloid-Leichtketten-Amyloidose (außer lokalisierte Amyloid-Leichtketten -Amyloidose, die auf die Haut oder das Knochenmark beschränkt ist)
- Plasmazell-Leukämie
- Vorherige Chemo- oder Strahlentherapie in den letzten 5 Jahren (außer lokaler Strahlentherapie bei lokalem Fortschreiten des Myeloms)
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Essen
Ev. Krankenhaus Essen-Werden gGmgH, Akademisches Lehrkrankenhaus der Univerität Duisburg/Essen, Hämatologie
0201-4089-0
Studien
GMMG-HD8/DSMM-XIX
Induktionstherapie mit Lenalidomid, Bortezomib und Dexamethason und entweder intravenösem oder subkutanem Isatuximab bei Patienten mit neu diagnostiziertem Multiplem Myelom, die für eine Hochdosis-Chemotherapie mit nachfolgender autologer Stammzelltransplantation geeignet sind
Organisatorische Daten:
| Prüfplancode: | GMMG-HD 8 / DSMM XIX |
| ISRCTN: | |
| EudraCT: | 2022-000996-38 |
| Clinicaltrials.gov: | NCT05804032 |
| DRKS: | |
| Sponsor: | Universitätsklinikum Heidelberg DSMM, Sanofi und KKS-Netzwerk |
| Studienphase: | |
| Status: | Rekrutierung geplant bis Juni 2025 |
Ziel:
In dieser Studie für Patienten mit zuvor unbehandeltem Multiplem Myelom soll die Wirksamkeit der Induktionstherapie mit Isatuximab in Kombination mit Lenalidomid/Bortezomib/Dexamethason (RVd) bei Gabe von Isatuximab unter die Haut (subkutan) gegenüber einer Gabe in die Vene (intravenös) untersucht werden.
Durchführung:
Randomisierung: Die Patienten werden vor der Induktionstherapie nach dem Zufallsverfahren (randomisiert) einem von 2 Studienarmen (A oder B) zugeordnet.
- Patienten in Arm A erhalten 3 Zyklen des monoklonalen Antikörpers Isatuximab intravenös in Kombination mit dem RVd-Schema (Lenalidomid, Bortezomib, Dexamethason). Jeder Zyklus hat eine Dauer von 42 Tagen.
- Patienten in Arm B erhalten 3 Zyklen Isatuximab subkutan in Kombination mit RVd.
Nach der Induktionstherapie erhalten die Patienten eine Standard-Intensivierung (in der Regel eine Mobilisierungstherapie auf Cyclophosphamid-Basis, Stammzellenentnahme und hochdosiertes Melphalan, gefolgt von einer autologen Stammzelltransplantation
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- Bestätigte Diagnose eines neu diagnostizierten behandlungsbedürftigen Multiplen Myeloms gemäß den überarbeiteten Diagnosekriterien der International Myeloma Working Group (IMWG))
- Der Patient ist für eine Hochdosis-Chemotherapie und eine autologe Stammzelltransplantation geeignet
- Messbare Krankheitsaktivität (gemäß der Definition im Prüfplan)
- Alter 18 bis 70 Jahre bei Studieneinschluss
Ausschlußkriterien:
- Bekannte Überempfindlichkeit (oder Kontraindikation) gegen einen der Bestandteile der Studientherapie
- Systemische Amyloid-Leichtketten-Amyloidose (außer lokalisierte Amyloid-Leichtketten -Amyloidose, die auf die Haut oder das Knochenmark beschränkt ist)
- Plasmazell-Leukämie
- Vorherige Chemo- oder Strahlentherapie in den letzten 5 Jahren (außer lokaler Strahlentherapie bei lokalem Fortschreiten des Myeloms)
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Klinikum Essen-Süd, Akademisches Lehrkrankenhaus der Univerität Duisburg/Essen, Hämatologie
0201-4089-2233
Universitätsklinik Essen, Zentrum für Innere Medizin, Klinik für Hämatologie
0201-723-0
Studien
M22-947 (ABBV-383)Intravenös verabreichtes ABBV-383 in Kombination mit Krebstherapien bei Patienten mit rezidiviertem oder refraktärem Multiplen Myelom: eine Studie zur Bewertung der unerwünschten Ereignisse und der Veränderung der Krankheitsaktivität
Organisatorische Daten:
NCT05259839
| Prüfplancode: |
M22-947 |
| ISRCTN: | |
| EudraCT: | |
| Clinicaltrials.gov: | NCT05259839 |
| DRKS: | |
| Sponsor: | TeneoOne Inc |
| Studienphase: | Phase 1, Erstanwendung am Menschen |
| Status: | Rekrutierung läuft, geplant bis Okt.2028 |
Ziele:
Das Multiple Myelom (MM) ist eine Plasmazellerkrankung, die durch das Wachstum klonaler Plasmazellen im Knochenmark gekennzeichnet ist. Ziel dieser Studie ist es, die Sicherheit und Toxizität von ABBV-383 bei gleichzeitiger Verabreichung mit Pomalidomid-Dexamethason (Pd), Lenalidomid-Dexamethason (Rd), Daratumumab-Dexamethason (Dd) oder Nirogacestat (Niro) bei erwachsenen Teilnehmern mit rezidiviertem/refraktärem Multiplem Myelom (MM) zu untersuchen. Es werden unerwünschte Ereignisse und Veränderungen der Krankheitsaktivität bewertet.
Durchführung:
In dieser Studie werden die Teilnehmer in Gruppen eingeteilt, die als Behandlungsarme bezeichnet werden. Untersucht wird ABBV-383 in Kombination mit Pd, Rd, Dd oder Niro. Jeder Behandlungsarm erhält eine andere Behandlungskombination, je nach Stadium der Studie und Eignung des Teilnehmers. Die Studie umfasst eine Dosissteigerungsphase, in der die beste Dosis von ABB-383 ermittelt werden soll. Anschließend folgt eine Dosiserweiterungsphase, in der die Dosis bestätigt werden soll.. Ungefähr 270 erwachsene Teilnehmer mit rezidiviertem/refraktärem MM werden an etwa 45 Standorten weltweit in die Studie aufgenommen. Die Teilnehmer erhalten ABBV-383 intravenös (i.v.) in Kombination mit oral/i.v. verabreichtem Pd, oral/i.v. verabreichtem Rd, oral/i.v./subkutan (s.c.) verabreichtem Dd oder oral/i.v. verabreichtem Niro in 21-tägigen Zyklen.
Für die Teilnehmer an dieser Studie kann die Belastung durch die Behandlung höher sein als bei ihrer Standardbehandlung. Während der Studie werden die Teilnehmer regelmäßig untersucht und die Wirkung und etwaige Nebenwirkungen der Behandlung werden häufig durch medizinische Beurteilungen und Blutuntersuchungen und Fragebögen überprüft.
Hintergrund:
ABBV-383 ist ein Prüfpräparat, das für die Behandlung des rezidivierenden/refraktären Multiplen Myeloms entwickelt wird. Es ist ein Antikörper, der (wie Elranatamab und Teclistamab) gegen zwei Ziele gerichtet (bispezifisch) ist: gegen BCMA und gegen CD3. BCMA ist ein Zelloberflächenprotein, das besonders auf Myelom- und Plasmazellen vorkommt und eine besondere biologische Bedeutung für das Überleben der Plasmazelle hat. Es wird bei Menschen mit Multiplem Myelom in signifikant höherem Maße exprimiert. CD3 ist an der Aktivierung der Immunantwort zur Bekämpfung von Infektionen beteiligt.
.
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- Dokumentierte Diagnose eines Multiplen Myeloms gemäß den IMWG-Diagnosekriterien
- ECOG (Index zur Abstufung der Lebensqualität von Patienten mit Krebserkrankungen) von 0, 1 oder2
- Es muss eine bestätigte Diagnose eines rezidivierten/refraktären Multiplen Myeloms (MM) mit dokumentierten Hinweisen auf ein Fortschreiten der Erkrankung während oder nach der letzten Behandlung des Teilnehmers vorliegen, wie vom Prüfarzt anhand der Kriterien der International Myeloma Working Group (IMWG) ermittelt wurde.
- Es muss eine messbare Erkrankung vorliegen, wie im Prüfplan dargelegt.
- Teilnehmer sind bislang nicht mit ABBV-383 vorbehandelt und haben noch nie eine BCMA-gerichtete Therapie erhalten. Teilnehmer, die eine zielgerichtete Therapie gegen Nicht-BCMA-Targets erhalten haben, werden nicht ausgeschlossen.
Ausschlusskriterien:
- Periphere autologe Stammzelltransplantation (SZT) innerhalb von 12 Wochen oder allogene SZT innerhalb eines Jahres vor der ersten Gabe des Studienmedikaments.
- Fortbestehende unerwünschte Ereignisse ≥ Grad 2 (National Cancer Institute [NCI] Common Terminology Criteria for Adverse Events [CTCAE] Version 5.0) infolge einer früheren Krebstherapie.
- Bekannter Myelombefall des zentralen Nervensystems.
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt
Erstanwendung von REGN5458 am Menschen bei Patienten mit rezidiviertem oder refraktärem Multiplem Myelom (MM)
Organisatorische Daten:
| Prüfplancode: | R5458-ONC-1826 |
| ISRCTN: | |
| EudraCT: | 2018-003188-78 |
| Clinicaltrials.gov: | NCT03761108 |
| DRKS: | |
| Sponsor: | Regeneron Pharmaceuticals |
| Studienphase: | Phase 1/2 Erstanwendung am Menschen |
| Status: | Rekrutierung läuft, geplant bis Mai 2025 |
Ziele:
Im Phase-1-Teil der Studie: Bewertung der Sicherheit, Verträglichkeit und dosisbegrenzender Nebenwirkungen (dosislimitierender Toxizitäten) und Ermittlung eines für die Phase 2 empfohlenen Dosierungsschemas von REGN5458 als Monotherapie bei Patienten mit rezidiviertem oder refraktärem multiplem Myelom.
Im Phase-2-Teil der Studie: Bewertung der Anti-Tumor-Aktivität von REGN5458, gemessen an der objektiven Ansprechrate und bestimmt durch ein unabhängiges Prüfungskomitee.
Hintergrund:
REGN5458 ist ein Antikörper, der gegen zwei Ziele gerichtet (bispezifisch) ist. Er soll an das Oberflächenprotein BCMA (B-cell maturation antigen) auf multiplen Myelomzellen und an den CD3-Rezeptor auf T-Zellen binden, um sie miteinander zu verbinden und die T-Zellen zur Abtötung der Krebszellen zu aktivieren.
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- ECOG Performance Status (Index zur Abstufung der Lebensqualität von Patienten mit Krebserkrankungen) von 0 oder 1.
- Bestätigte Diagnose eines aktiven Multiplen Myeloms gemäß den von der International Myeloma Working Group (IMWG) aufgestellten Diagnosekriterien
- Das Ansprechen muss anhand der Ansprechkriterien der IMWG auswertbar sein.
-
Phase 1 Dosissteigerung:
- Patienten mit MM, die alle Behandlungsmöglichkeiten ausgeschöpft haben, von denen ein bedeutsamer klinischer Nutzen zu erwarten ist, was sich entweder durch einen Krankheitsrückfall, eine behandlungsrefraktäre Erkrankung oder eine Unverträglichkeit der Therapie zeigt und einen der folgenden Punkte umfasst:
- Fortschreiten der Erkrankung während oder nach mindestens drei Therapielinien oder Unverträglichkeit der Therapie, einschließlich eines Proteasom-Inhibitors, eines immunmodulatorischen Wirkstoffs (IMiD) und eines Anti-CD38-Antikörpers ODER
- Fortschreiten der Krankheit während oder nach einem Anti-CD38-Antikörper und „doppelte Refraktärität“ gegenüber einem Proteasom-Inhibitor und einem IMiD oder Unverträglichkeit der Therapie. Der Anti-CD38-Antikörper kann allein oder in Kombination mit einem anderen Wirkstoff, z. B. einem Proteasom-Inhibitor, verabreicht worden sein. Eine refraktäre Erkrankung ist definiert als Ausbleiben des Ansprechens oder Rückfall innerhalb von 60 Tagen nach der letzten Behandlung.
Phase 2:
- Die Patienten müssen dreifach refraktär sein, d. h. sie müssen auf eine vorherige Behandlung mit mindestens einem Anti-CD38-Antikörper, einem Proteasom-Inhibitor und einem IMiD refraktär sein. Darüber hinaus müssen die Patienten fünf Wirkstoffe erhalten haben (d. h. sie müssen zuvor mit zwei Proteasom-Inhibitoren, zwei IMiDs [Lenalidomid und Pomalidomid] und einem monoklonalen Anti-CD38-Antikörper behandelt worden sein).
Ausschlusskriterien:
- Diagnose einer Plasmazellleukämie, einer primären systemischen Leichtketten-Amyloidose (ausgenommen Myelom-assoziierte Amyloidose), einer Waldenström-Makroglobulinämie (lymphoplasmatisches Lymphom) oder eines POEMS-Syndroms (Polyneuropathie, Organomegalie, Endokrinopathie, monoklonale Proteine und Hautveränderungen)
- Patienten mit bekannten MM-Hirnläsionen oder meningealer Beteiligung
- Vorherige Behandlung mit BCMA-gerichteten Immuntherapien, einschließlich BCMA-bispezifischer Antikörper und BiTEs sowie BCMA-CAR-T-Zellen.
- Hinweis: BCMA-Antikörper-Wirkstoff-Konjugate sind nicht ausgeschlossen
- Vorgeschichte einer allogenen Stammzelltransplantation zu irgendeinem Zeitpunkt oder einer autologen Stammzelltransplantation innerhalb von 12 Wochen vor Beginn der Studienbehandlung
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt
Goch
Katholisches Karl-Leisner-Klinikum gGmbH - Wilhelm-Anton-Hospital Klinik für Innere Medizin
02823 - 891-447
Hagen
Kathol. Krankenhaus Hagen, Abt. Hämatologie/Onkologie
02331-129-250
Studien
GMMG-HD8/DSMM-XIX
Induktionstherapie mit Lenalidomid, Bortezomib und Dexamethason und entweder intravenösem oder subkutanem Isatuximab bei Patienten mit neu diagnostiziertem Multiplem Myelom, die für eine Hochdosis-Chemotherapie mit nachfolgender autologer Stammzelltransplantation geeignet sind
Organisatorische Daten:
| Prüfplancode: | GMMG-HD 8 / DSMM XIX |
| ISRCTN: | |
| EudraCT: | 2022-000996-38 |
| Clinicaltrials.gov: | NCT05804032 |
| DRKS: | |
| Sponsor: | Universitätsklinikum Heidelberg DSMM, Sanofi und KKS-Netzwerk |
| Studienphase: | |
| Status: | Rekrutierung geplant bis Juni 2025 |
Ziel:
In dieser Studie für Patienten mit zuvor unbehandeltem Multiplem Myelom soll die Wirksamkeit der Induktionstherapie mit Isatuximab in Kombination mit Lenalidomid/Bortezomib/Dexamethason (RVd) bei Gabe von Isatuximab unter die Haut (subkutan) gegenüber einer Gabe in die Vene (intravenös) untersucht werden.
Durchführung:
Randomisierung: Die Patienten werden vor der Induktionstherapie nach dem Zufallsverfahren (randomisiert) einem von 2 Studienarmen (A oder B) zugeordnet.
- Patienten in Arm A erhalten 3 Zyklen des monoklonalen Antikörpers Isatuximab intravenös in Kombination mit dem RVd-Schema (Lenalidomid, Bortezomib, Dexamethason). Jeder Zyklus hat eine Dauer von 42 Tagen.
- Patienten in Arm B erhalten 3 Zyklen Isatuximab subkutan in Kombination mit RVd.
Nach der Induktionstherapie erhalten die Patienten eine Standard-Intensivierung (in der Regel eine Mobilisierungstherapie auf Cyclophosphamid-Basis, Stammzellenentnahme und hochdosiertes Melphalan, gefolgt von einer autologen Stammzelltransplantation
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- Bestätigte Diagnose eines neu diagnostizierten behandlungsbedürftigen Multiplen Myeloms gemäß den überarbeiteten Diagnosekriterien der International Myeloma Working Group (IMWG))
- Der Patient ist für eine Hochdosis-Chemotherapie und eine autologe Stammzelltransplantation geeignet
- Messbare Krankheitsaktivität (gemäß der Definition im Prüfplan)
- Alter 18 bis 70 Jahre bei Studieneinschluss
Ausschlußkriterien:
- Bekannte Überempfindlichkeit (oder Kontraindikation) gegen einen der Bestandteile der Studientherapie
- Systemische Amyloid-Leichtketten-Amyloidose (außer lokalisierte Amyloid-Leichtketten -Amyloidose, die auf die Haut oder das Knochenmark beschränkt ist)
- Plasmazell-Leukämie
- Vorherige Chemo- oder Strahlentherapie in den letzten 5 Jahren (außer lokaler Strahlentherapie bei lokalem Fortschreiten des Myeloms)
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Hamm
Klinik für Hämatologie und Onkologie - St. Josef-Krankenhaus Hamm
02381 961-1951
Köln
Onkologische Gemeinschaftspraxis Köln
0221 - 931 822 0
Universitätsklinikum Köln Medizinische Klinik I
0221-478-96548
Studien
DREAMM 7Belantamab-Mafodotin
Bewertung der Wirksamkeit und Sicherheit von Belantamab-Mafodotin, Bortezomib und Dexamethason gegenüber Daratumumab, Bortezomib und Dexamethason bei Teilnehmern mit rezidiviertem/refraktärem Multiplen Myelom
Organisatorische Daten:
| Prüfplancode: | 207503 |
| ISRCTN: | |
| EudraCT: | 2018-003993-29 |
| Clinicaltrials.gov: | NCT04246047 |
| DRKS: | |
| Sponsor: | GlaxoSmithKline |
| Studienphase: | III |
| Status: | Rekrutierung geplant bis Juni/2026 |
Kurzbeschreibung:
Hierbei handelt es sich um eine randomisierte, offene Phase-3-Studie zur Untersuchung der Sicherheit und Wirksamkeit von Belantamab-Mafodotin in Kombination mit Bortezomib/Dexamethason (Arm A) gegenüber Daratumumab in Kombination mit Bortezomib/Dexamethason (Arm B) bei Teilnehmern mit rezidivierendem multiplem Myelom..
Hintergrund:
Die amerikanische Arzneimittelbehörde FDA hat Belantamab-Mafodotin-blmf (GSK2857916; Blenrep) für die Behandlung von Patienten mit rezidiviertem oder refraktärem Multiplen Myelom zugelassen, die zuvor mit mindestens 4 Therapien behandelt wurden, darunter ein immunmodulatorisches Mittel, ein Proteasom-Inhibitor und ein Anti-CD38-Antikörper. Dies ist weltweit die erste Anti-BCMA-Behandlung, die bei dieser Patientenpopulation verfügbar ist.
Belantamab-Mafodotin ist gegen das B-Zell-Reifungsantigen (BCMA) gerichtet, das eine wichtige Rolle für das Überleben von Plasmazellen spielt und auf Zellen des Multiplen Myeloms exprimiert wird. Das Antikörper-Wirkstoff-Konjugat Belantamab-Mafodotin bindet an BCMA auf Myelom-Zelloberflächen, wodurch der Zellzyklus angehalten und eine antikörperabhängige zelluläre Zytotoxizität induziert wird.
Normalerweise fördert BCMA das Überleben von Plasmazellen durch Signalübertragung von zwei bekannten Liganden. Dieser Signalweg hat sich als wichtig für das Wachstum und Überleben von Myelomzellen erwiesen.
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- Bestätigte Diagnose eines Multiplen Myeloms (MM) gemäß IMWG-Kriterien
- Vorbehandlung des MM mit mindestens einer Therapielinie und dokumentierte Krankheitsprogression während oder nach der letzten Therapie.
- ECOG Performance Status (Index zur Abstufung der Lebensqualität von Patienten mit Krebserkrankungen) von 0 bis 2
- Muss mindestens 1 Aspekt einer messbaren Erkrankung aufweisen, wie folgt definiert:
- Urin-M-Protein-Ausscheidung > 200 mg pro 24 Stunden, oder
- Serum-M-Protein-Konzentration > 0,5 g/dl, oder
- Freie Leichtketten im Serum (FLC): beteiligter FLC-Spiegel > 10 mg/dl (> 100 mg/L) und ein abnormes Verhältnis der freien Leichtketten im Serum (< 0,26 oder > 1,65).
- Alle Nebenwirkungen, die infolge durch Vorbehandlungen hervorgerufen wurden, müssen zum Zeitpunkt der Rekrutierung < Grad 1 sein, mit Ausnahme von Alopezie.
Ausschlußkriterien:
- Unverträglichkeit gegenüber Daratumumab.
- Kein bzw. kein hinreichendes Ansprechen (therapierefraktär) gegenüber Daratumumab oder einer anderen Anti-CD38-Therapie (definiert als fortschreitende Erkrankung während der Behandlung mit einer Anti-CD38-Therapie oder innerhalb von 60 Tagen nach Abschluss dieser Behandlung).
- Unverträglichkeit gegenüber Bortezomib oder kein bzw. kein hinreichendes Ansprechen (therapierefraktär) gegenüber Bortezomib (definiert als fortschreitende Erkrankung während der Behandlung mit einem Bortezomib-haltigen Schema von 1,3 mg/m2 zweimal wöchentlich oder innerhalb von 60 Tagen nach Abschluss dieser Behandlung). Hinweis: Patienten mit fortschreitender Erkrankung während der Behandlung mit einem wöchentlichen Bortezomib-Schema dürfen teilnehmen.
- Anhaltende periphere Neuropathie von Grad 2 oder höher oder neuropathische Schmerzen.
- Vorherige Behandlung mit einer Anti-B-Zellreifungsantigen- (Anti-BCMA-) Therapie.
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Daratumumab zur Erstlinienbehandlung von Patienten mit Myelom, die für eine Transplantation nicht in Frage kommen, gefolgt von einer erneuten Daratumumab-Behandlung beim ersten Rückfall (Rezidiv)
.Organisatorische Daten:
| Prüfplancode: | Uni-Koeln-3946 |
| ISRCTN: | |
| EudraCT: | 2019-003856-35 |
| Clinicaltrials.gov: | NCT04656951 |
| DRKS: | |
| Sponsor: | Universitätsklinnikum Köln/Janssen |
| Studienphase: | Phase 2 |
| Status: | Rekrutierung läuft , bis Dez 2029 |
Kurzbeschreibung
Dies ist eine Studie zur Bewertung der Wirksamkeit und Sicherheit des Medikaments Daratumab bei Patienten mit unbehandeltem Multiplem Myelom (MM).
Untersucht wird:
1. die Sicherheit und Wirksamkeit von Daratumumab als Zusatz zu einem Standardinduktionsregimes mit Bortezomib, Cyclophosphamid und Dexamethason (VCD)
2. die Wirksamkeit der Erhaltungstherapie bis zur Progression unter Anwendung von Daratumumab in Kombination mit Bortezomib und Dexamethason
3. die Wirksamkeit eines Daratumumab-haltigen Regimes beim ersten Rezidiv nach einem Daratumumab-haltigen Erstlinienregime
Hintergrund:
Daratumumab ist ein monoklonaler Antikörper, der zur Behandlung des Multiplen Myeloms eingesetzt wird. Der Antikörper bindet an das Oberflächenprotein CD38, das von den Tumorzellen in allen Stadien der Erkrankung gebildet wird. Die Bindung des Antikörpers führt zum Absterben der Tumorzelle durch Apoptose sowie durch eine verstärkte Immunreaktion.
VCD (Velcade, Cyclophosphamid, Dexamethason) hat sich in Deutschland als Standard für die Erstlinientherapie von Patienten etabliert, die für eine Transplantation in Frage kommen. Aufgrund des günstigen Nebenwirkungsprofils ist dieses Regime auch für Patienten geeignet, die für eine Transplantation nicht in Frage kommen. Daratumumab (Dara) hat sich in einer Vielzahl von Kombinationen wie Bortezomib/Melphalan/Prednison (VMP), Bortezomib/Dexamethason (Vd) oder Lenalidomid/Dexamethason (Rd) als sicher und wirksam erwiesen. Unter Verwendung einer reduzierten Cyclophosphamid-Dosis soll in der Studie die Sicherheit und Wirksamkeit von Dara in Kombination mit VCD (Dara-VCD) bei Patienten untersucht werden, die für eine Transplantation nicht in Frage kommen.
Die Erhaltungstherapie ist bei Patienten, die für eine Transplantation in Frage kommen, und bei Patienten, die nicht dafür in Frage kommen, zum Standard geworden. Es besteht jedoch Unsicherheit darüber, ob eine Erhaltungstherapie mit einem einzigen Wirkstoff ausreicht oder ob eine Kombinationstherapie erforderlich ist. In dieser Studie wird die Sicherheit und Wirksamkeit der Erhaltungstherapie mit Daratumumab in Kombination mit Bortezomib und Dexamethason untersucht.
Die Vielzahl der Behandlungsmöglichkeiten beim ersten Rückfall hat dazu geführt, dass eine optimale Abfolge von Therapieschemata in der Erst- und Zweitlinie gefunden werden muss. Insbesondere ist unklar, ob die Wirksamkeit von Daratumumab bei einem Rückfall beeinflusst wird, wenn es vorher als Teil der Erstlinienbehandlung verwendet wurde.
Einschlusskriterien:
- Patienten müssen ein unbehandeltes, dokumentiertes, behandlungsbedürftiges multiples Myelom haben, das folgenden Kriterien erfüllt:
- Monoklonale Plasmazellen im Knochenmark ≥ 10% und/oder Nachweis eines Plasmozytoms, nachgewiesen durch eine Biopsie
- Multiples Myelom IgG: Serumspiegel des monoklonalem Paraproteins (M-Protein) ≥ 1,0 g/dl oder M-Proteinspiegel im Urin ≥ 200 mg/24 Stunden; oder
- Multiples Myelom mit IgA, IgD, IgE, IgM: M-Proteinspiegel im Serum ≥ 0,5 g/dl oder M-Proteinspiegel im Urin ≥ 200 mg/24 Stunden; oder
- Multiples Leichtketten-Myelom ohne messbare Erkrankung im Serum oder Urin: Freie Leichtketten-Immunoglobuline im Serum ≥ 10 mg/dl und abnormales Verhältnis der freien Leichtketten-Immunoglobuline Kappa Lambda.
- ECOG < 2 (Index zur Abstufung der Lebensqualität von Patienten mit Krebserkrankungen)
Ausschlusskriterien:
- Vorherige Therapie des multiplen Myeloms, mit Ausnahme von Dexamethason in einer Höchstdosis von insgesamt 160 mg, einer Notfall-Strahlentherapie oder einer Operation zur Linderung der Symptome
- Teilnahme an anderen interventionellen klinischen Studien
- bekannte meningeale Beteiligung des Multiplen Myeloms.
- Vorgeschichte mit einer bösartigen Erkrankung (außer dem Multiplen Myelom) innerhalb von 3 Jahren vor dem Datum des Screenings.
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt
CAR T-Zelltherapie mit bb2121
Eine Phase-2-Studie zur Bestimmung der Wirksamkeit und Sicherheit von bb2121 bei Patienten mit rezidiviertem (wieder aufgetretenem) und refraktärem (nicht auf Behandlung ansprechendem) Multiplem Myelom (RRMM) und bei Patienten mit risikoreichem Multiplem Myelom, bei denen die Krankheit innerhalb von 18 Monaten nach der Erstbehandlung fortgeschritten ist.
Organisatorische Daten:
| Prüfplancode: | KarMMa2 |
| ISRCTN: | |
| EudraCT: | 2018-000264-28 |
| Clinicaltrials.gov: | NCT03601078 |
| DRKS: | |
| Sponsor: | Celgene |
| Studienphase: | Phase 2 |
| Status: | Rekrutierung läuft, geplant bis März 2025 |
Ziel:
In dieser Studie soll die Wirksamkeit und Sicherheit von bb2121 bei Patienten mit rezidiviertem oder refraktären Multiplen Myelom bewertet werden.
Hintergrund:
Die chimäre-Antigenrezeptor-(CAR) T-Zelltherapie bb2121 ist gegen das Oberflächenantigen BCMA (engl. B-cell maturation antigen) gerichtet, um T-Zellen dahin gehend zu programmieren, dass sie maligne Myelomzellen erkennen und abtöten. https://meetinglibrary.asco.org/record/160693/abstract
In einer Phase-1-Studie bei stark vorbehandelten Patienten mit rezidiviertem oder refraktären multiplem Myelom zeigte bb2121 ermutigende Ergebnisse mit tiefem und dauerhaftem Ansprechen bei gleichzeitig guter Verträglichkeit. https://www.presseportal.de/pm/62626/3972298 In der vorliegenden Studie soll die Wirksamkeit und Sicherheit von bb2121 als mögliche Therapie für Multiples Myelom weiter untersucht werden.
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- Es besteht eine messbare Krankheit, definiert als
- M-Protein (Serum-Protein-Elektrophorese [sPEP] oder Urin-Protein-Elektrophorese [uPEP]): sPEP ≥ 0,5 g/dl oder uPEP ≥ 200 mg/24 Stunden und/oder
- Leichtketten-MM ohne messbare Krankheit im Serum oder Urin: freie Leichtketten im Serum ≥ 10 mg/dl und abnormes Kappa-Lambda-Leichtketten-Verhältnis im Serum
- Spezifische Anforderungen für:
- Kohorte 1: ≥ 3 frühere Anti-Myelom-Behandlungsschemen
- Kohorte 2: 1 frühere Anti-Myelom-Behandlung
- ECOG Performance Status von 0 oder 1 ((Index zur Abstufung der Lebensqualität von Patienten mit Krebserkrankungen)
Ausschlusskriterien:
- Erhalt eines Prüfpräparats innerhalb von 14 Tagen vor der Leukapherese
- Eines der folgenden Verfahren innerhalb der letzten 14 Tage vor der Leukapherese:
- Plasmapherese
- Große Operation (nach Definition des Prüfarztes)
- Strahlentherapie mit Ausnahme der lokalen Therapie für myelombedingten Knochenläsionen
- Anwendung einer systemischen Anti-Myelom-Medikamententherapie
- Bekannte Beteiligung des zentralen Nervensystems
- Klinischer Nachweis einer Lungenleukostase und Verbrauchskoagulopathie (disseminierten intravasale Gerinnung)
- Vorgeschichte oder Vorliegen einer klinisch relevanten Erkrankung des Zentralnervensystems (ZNS)
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Ergänzende Informationen
Zur Herstellung der mit einem bb2121 chimären Antigenrezeptor (CAR) modifizierten T-Zellen werden die weißen Blutkörperchen (Leukozyten) aus dem Blut herausgelöst. Dies bezeichnet man als Leukapherese. Vor der bb2121-Infusion wird bei den Patienten ein Verfahren zur Entfernung der Lymphozyten, die sogenannte Lymphozytendepletion, mit Fludarabin und Cyclophosphamid durchgeführt.
CAR T-Zelltherapie mit bb2121
Es handelt sich um eine multizentrische, randomisierte, offene Phase-3-Studie, in der die Wirksamkeit und Sicherheit von bb2121 im Vergleich zu den Standardschemata bei Patienten mit rezidiviertem und refraktärem multiplem Myelom (RRMM) verglichen wird.
Organisatorische Daten:
| Prüfplancode: | KarMMa-3 |
| ISRCTN: | U1111-1217-9988 |
| EudraCT: | 2018-001023-38 |
| Clinicaltrials.gov: | NCT03651128 |
| DRKS: | |
| Sponsor: | Celgene |
| Studienphase: | Phase 3 |
| Status: | Rekrutierung läuft, geplant bis Juni 2025 |
Ziel:
In dieser Studie soll die Wirksamkeit und Sicherheit von bb2121 bei Patienten mit rezidiviertem oder refraktären Multiplen Myelom im Vergleich zu einer Standardtherapie bewertet werden.
Hintergrund:
Die chimäre-Antigenrezeptor-(CAR) T-Zelltherapie bb2121 ist gegen das Oberflächenantigen BCMA (engl. B-cell maturation antigen) gerichtet, um T-Zellen dahin gehend zu programmieren, dass sie maligne Myelomzellen erkennen und abtöten. https://meetinglibrary.asco.org/record/160693/abstract
In einer Phase-1-Studie bei stark vorbehandelten Patienten mit rezidiviertem oder refraktärem Multiplem Myelom zeigte bb2121 ermutigende Ergebnisse mit tiefem und dauerhaftem Ansprechen bei gleichzeitig guter Verträglichkeit. https://www.presseportal.de/pm/62626/3972298
In der vorliegenden Studie soll die Wirksamkeit und Sicherheit von bb2121 als mögliche Therapie für Multiples Myelom im Vergleich zu einer Standardbehandlung weiter untersucht werden.
Durchführung
 Es wird erwartet, dass in der Studie etwa 381 Patienten mit RRMM randomisiert werden. Etwa 254 Patienten werden in Behandlungsarm A und etwa 127 Patienten in Behandlungsarm B randomisiert.
Es wird erwartet, dass in der Studie etwa 381 Patienten mit RRMM randomisiert werden. Etwa 254 Patienten werden in Behandlungsarm A und etwa 127 Patienten in Behandlungsarm B randomisiert.
Zur Herstellung der mit einem bb2121 chimären Antigenrezeptor (CAR) modifizierten T-Zellen werden die weißen Blutkörperchen (Leukozyten) aus dem Blut herausgelöst. Dies bezeichnet man als Leukapherese. Die Leukozyten werden dann behandelt und zurückinfundiert. Vor der bb2121-Infusion wird bei den Patienten ein Verfahren zur Entfernung der Lymphozyten, die sogenannte Lymphozytendepletion, mit Fludarabin und Cyclophosphamid durchgeführt.
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- Patient hat eine messbare Krankheit, definiert als:
- M-Protein (Serum-Protein-Elektrophorese [sPEP] oder Urin-Protein-Elektrophorese [uPEP]): sPEP ≥ 0,5 g/dl oder uPEP ≥ 200 mg/24 Stunden und/oderLeichtketten-MM ohne messbare Krankheit im Serum oder Urin: immunglobulinfreie Leichtketten im Serum ≥ 10 mg/dl und abnormes Kappa-Lambda-Leichtketten-Verhältnis im Serum
- Patient hat mindestens 2, aber höchstens 4 vorherige Behandlungsschemen für MM erhalten.
- Patient hat mindestens 2 Behandlungszyklen mit DARA, einem Proteasom-Inhibitor- und einem immunmodulatorischen Wirkstoffschema für mindestens 2 aufeinanderfolgende Zyklen erhalten.
- Patient muss gegenüber dem letzten Behandlungsschema refraktär sein, also ein dokumentiertes Fortschreiten der Erkrankung während oder innerhalb von 60 Tagen nach Abschluss des letzten Anti-Myelom-Schemas vor Studieneinschluss aufweisen.
- ECOG Performance Status von 0 oder 1 (Index zur Abstufung der Lebensqualität von Patienten mit Krebserkrankungen
Ausschlusskriterien:
- Nichtsekretorisches multiples Myelom (MM)
- Bestimmte auffällige Laborwerte
- unzureichende Lungenfunktion, definiert als eine Sauerstoffsättigung von weniger als 92 % bei Raumluft
- Maligne Erkrankungen neben MM in der Vorgeschichte. Dieses Kriterium gilt nicht, wenn diese Erkrankungen nicht in den letzten 5 Jahren oder länger vorlagen
- Bekannte Beteiligung des zentralen Nervensystems
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Studie mit Elranatamab im Vergleich zu Lenalidomid bei Patienten mit neu diagnostiziertem Multiplem Myelom nach Transplantation
Organisatorische Daten:
| Prüfplancode: | C1071005, MagnetisMM-7 |
| ISRCTN: | |
| EudraCT: | 2021-006052-14 |
| Clinicaltrials.gov: | NCT05317416 |
| DRKS: | |
| Sponsor: | Pfizer |
| Studienphase: | Phase 3 |
| Status: | Rekrutierung läuft, geplant bis Juni 2027 |
Randomisierte, 2-armige Phase-3-Studie mit Elranatamab (PF-06863135) im Vergleich zu Lenalidomid bei Patienten mit neu diagnostiziertem Multiplem Myelom, die nach einer autologen Stammzelltransplantation eine minimale Resterkrankung aufweisen.
Ziel:
Mit dieser Studie soll untersucht werden, ob eine Elranatamab-Monotherapie im Vergleich zu einer Lenalidomid-Monotherapie Teilnehmern mit neu diagnostiziertem Multiplem Myelom einen klinischen Nutzen bietet, bei denen nach einer autologen Stammzelltransplantation noch eine minimale Resterkrankung nachweisbar ist.
Hintergrund:
Der bispezifische BCMA/CD3-Antikörper Elranatamab (PF-06863135) zeigte bei Patienten mit rezidiviertem/refraktärem Multiplem Myelom eine vielversprechende vorläufige Wirksamkeit und Verträglichkeit.
Daten aus der Phase-1-Studie MagnetisMM-1 (NCT03269136), die auf der ASCO-Jahrestagung 2021 vorgestellt wurden, zeigten, dass Elranatamab in Dosen von bis zu 1000 µg/kg ein überschaubares Sicherheitsprofil ohne dosislimitierende Toxizitäten aufweist.
Darüber hinaus erzielte der Wirkstoff bei Dosierungen von 215 µg/kg und höher eine Gesamtansprechrate (ORR) von 70 % und eine vollständige Ansprechrate (CR)/stringente CR von 30 %. Bei der empfohlenen Phase-2-Dosis von 1000 µg/kg erzielte der Wirkstoff eine ORR von 83,3 %. Bemerkenswert ist, dass drei von vier in die Studie aufgenommenen Patienten, die zuvor mit einer BCMA-gerichteten Therapie behandelt worden waren, ein Ansprechen erzielten.
Durchführung:
Die Teilnehmer an der Studie werden nach dem Zufallsprinzip einer von zwei Behandlungsgruppen zuteilt.
- Teilnehmer in Gruppe 1 erhalten Elrantamab als Injektion unter die Haut in der Studienklinik. Nach den anfangs an den gegenwärtigen Zustand des Patienten angepassten Dosen (sogenannte Step-up-Dosen) erhalten die Teilnehmer dann eine Dosis Elrantamab pro Woche. Nach 6 Monaten wird die Häufigkeit der Klinikbesuche für die Verabreichung der Injektionen auf jede zweite Woche reduziert.
- Teilnehmer in Gruppe 2 erhalten Lenalidomid oral einmal täglich zu Hause.
Die Teilnahme an der Studie dauert etwa fünf Jahre.
Ein und Ausschlusskriterien:
Einschlusskriterien:
-
Diagnose eines Multiplen Myeloms gemäß den von der International Myeloma Working Group (IMWG) aufgestellten Diagnosekriterien
-
Vorgeschichte einer Induktionstherapie für neu diagnostiziertes MM, gefolgt von einer Hochdosistherapie und einer autologen Stammzelltransplantation (ASZT). Die Randomisierung muss innerhalb von 120 Tagen nach der ASZT erfolgen. Bei Teilnehmern, die nach der ASZT eine Konsolidierungstherapie erhalten, muss die Randomisierung innerhalb von 60 Tagen nach der Konsolidierung und innerhalb von 6 Monaten nach der ASZT erfolgen.
-
Partielles Ansprechen oder besser nach den IMWG-Kriterien zum Zeitpunkt der RandomisierungNachweis einer minimalen Resterkrankung (MRD-positiv, ≥ 10-5) beim Screening.
-
ECOG Performance Status (Index zur Abstufung der Lebensqualität von Patienten mit Krebserkrankungen) von 0 oder 1.
Ausschlusskriterien:
-
Plasmazell-Leukämie
-
POEMS-Syndrom
-
Amyloidose, Waldenström-Makrogobulinämie
-
Bekannte aktive Beteiligung des ZNS oder klinische Anzeichen einer Beteiligung der Hirnhäute (Meningen)
-
Vorherige MM-Erhaltungstherapie
-
Vorherige Behandlung mit einer BCMA-Therapie
-
Jede andere aktive bösartige Erkrankung innerhalb von 3 Jahren vor der Aufnahme in die Studie, mit Ausnahme von adäquat behandeltem Basalzell- oder Plattenepithelkarzinom der Haut oder Karzinom in situ (z. B. bei der Diagnose) oder vor der Transplantation.
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt
Talquetamab
Talquetamab in Kombination mit Daratumumab oder in Kombination mit Daratumumab und Pomalidomid im Vergleich zu Daratumumab in Kombination mit Pomalidomid und Dexamethason bei Teilnehmern mit rezidiviertem oder refraktärem Multiplem Myelom (MonumenTAL-3)
Organisatorische Daten:
| Prüfplancode: | CR109082, 64407564MMY3002 |
| ISRCTN: | |
| EudraCT: | 2021-000202-22 |
| Clinicaltrials.gov: | NCT05455320 |
| DRKS: | |
| Sponsor: | Janssen |
| Studienphase: | 3 |
| Status: | Rekrutierung geplant bis Febr 2029 |
Ziel:
Ziel der Studie ist der Vergleich der Wirksamkeit von unter die Haut (subkutan, s.c.) verabreichtem Talquetamab in Kombination mit Daratumumab s.c. und Pomalidomid (Tal-DP) bzw. Talquetamab s.c. in Kombination mit Daratumumab s.c. (Tal-D) gegenüber Daratumumab s.c. in Kombination mit Pomalidomid und Dexamethason (DPd) bei Teilnehmern mit rezidiviertem oder refraktärem Multiplem Myelom, die mindestens eine vorherige Therapielinie erhalten haben.
Hintergrund:
Talquetamab ist ein humanisierter monoklonaler Antikörper, der gegen zwei Ziele gerichtet (bispezifisch) ist: gegen humanes CD3, ein Oberflächenantigen auf T-Zellen, und gegen GPRC5D (G protein-coupled receptor family C group 5 member D), ein Antigen in Tumorzellen oder auf deren Oberfläche (tumor-assoziiertes Antigen). Bei Verabreichung bindet Talquetamab sowohl an CD3 auf T-Zellen als auch an GPRC5D, das auf bestimmten Tumorzellen wie z.B. dem Multiplen Myelom vermehrt vorkommt und eine Schlüsselrolle bei der Vermehrung von Tumorzellen spielt.
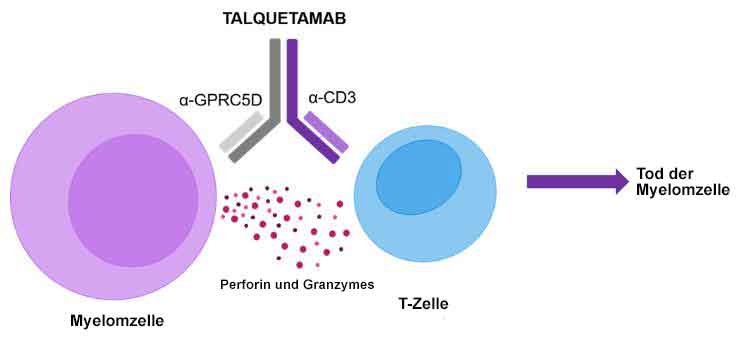
Abb. aus https://multiplemyelomahub.com/,
Durchführung:
Die Studie ist in drei Phasen unterteilt: Voruntersuchung (Screening), Behandlung (bis eines der folgenden Ereignisse eintritt: bestätigtes Fortschreiten der Krankheit, Tod, unverträgliche Toxizität, Widerruf der Einwilligung oder Ende der Studie) und Nachbeobachtung (bis eines der folgenden Ereignisse eintritt: Tod, Widerruf der Einwilligung, Verlust der Nachbeobachtung oder Ende der Studie). Wirksamkeit, Sicherheit, Pharmakokinetik, Immunogenität und Biomarker werden zu bestimmten Zeitpunkten untersucht. Die Gesamtdauer der Studie beträgt bis zu 6 Jahren und 6 Monaten.
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- Dokumentiertes Multiples Myelom, definiert als
- a) Diagnose des Multiplen Myeloms gemäß den diagnostischen Kriterien der International Myeloma Working Group (IMWG) und
- b) messbare Erkrankung zum Zeitpunkt des Screenings gemäß der Definition im Protokoll
- Rezidivierte oder refraktäre Erkrankung gemäß folgender Definition:
- eine rezidivierte Erkrankung ist definiert als anfängliches Ansprechen auf die vorherige Behandlung, gefolgt von einer nach IMWG-Kriterien bestätigten fortschreitenden (progredienten) Erkrankung mehr als 60 Tage nach Beendigung der Behandlung;
- eine refraktäre Erkrankung ist definiert als eine Verringerung des monoklonalen Paraproteins (M-Protein) um weniger als 25 % oder eine nach IMWG-Kriterien bestätigte fortschreitende Erkrankung während der vorherigen Behandlung oder ≤ 60 Tage nach Beendigung der Behandlung
- Teilnehmer müssen mindestens eine vorherige Linie einer Myelom-Therapie, einschließlich eines Proteasom-Inhibitors (PI) und Lenalidomid, erhalten haben. Patienten, die nur eine vorherige Linie einer Myelom-Therapie erhalten haben, müssen als Lenalidomid-refraktär gelten (d. h. bei ihnen ist bei Beendigung einer Lenalidomid-haltigen Therapie oder innerhalb von 60 Tagen danach eine fortschreitende (progrediente) Erkrankung nach IMWG-Kriterien nachweisbar). Teilnehmer, die 2 oder mehr vorherige Therapielinien zur Behandlung des Myeloms erhalten haben, müssen als Lenalidomid-behandelt gelten
- Dokumentierte Anzeichen einer fortschreitenden Erkrankung, basierend auf der Feststellung des Ansprechens durch den Prüfarzt gemäß den IMWG-Kriterien während oder nach der letzten Behandlung
- ECOG (Index zur Abstufung der Lebensqualität von Patienten mit Krebserkrankungen) von 0, 1 oder 2
Ausschlußkriterien
- Gegenanzeigen oder lebensbedrohliche Allergien, Überempfindlichkeiten oder Unverträglichkeiten gegenüber Hilfsstoffen des Studienmedikaments
- Refraktär gegenüber einem monoklonalen Antikörper gegen Cluster of Differentiation 38 (CD38), wie in den IMWG-Konsensrichtlinien definiert (Fortschreiten der Erkrankung während der Behandlung oder innerhalb von 60 Tagen nach Beendigung der Therapie mit einem monoklonalen Antikörper gegen CD38)
- Erhalt von Kortikosteroiden in einer maximalen Gesamtdosis von oder entsprechend ≥ 140 mg Prednison innerhalb von 14 Tagen vor Gabe der ersten Dosis des Studienmedikaments
- Bekannte aktive Beteiligung des zentralen Nervensystems (ZNS) oder klinische Anzeichen einer meningealen Beteiligung des Multiplen Myeloms. Bei Verdacht auf eine der beiden Erkrankungen sind eine negative Magnetresonanztomographie (MRT) des gesamten Gehirns und eine lumbale Zytologie erforderlich.
- Plasmazellleukämie zum Zeitpunkt der Voruntersuchung, Waldenström-Makroglobulinämie, Polyneuropathie, Organomegalie, Endokrinopathie, M-Protein und Hautveränderungen (POEMS-Syndrom) oder primäre Amyloid-Leichtketten-Amyloidose
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Venetoclax in Kombination mit Daratumumab /Dexamethason
Eine Studie zur Kombinationstherapie mit Venetoclax, Daratumumab und Dexamethason (mit und ohne Bortezomib) bei Patienten mit rezidiviertem oder refraktärem Multiplem Myelom.
Organisatorische Daten:
| Prüfplancode: | M15-654 |
| ISRCTN: | |
| EudraCT: | 2017-002099-26 |
| Clinicaltrials.gov: | NCT03314181 |
| DRKS: | |
| Sponsor: | AbbVie in Zusammenarbeit mit Janssen Research & Development, LLC |
| Studienphase: | Phase 1/2 |
| Status: | Rekrutierung läuft, geplant bis 6/2024 |
Kurzbeschreibung:
Multizentrische Dosissteigerungs- und Erweiterungsstudie der Phase 1 / 2 zur Bewertung der Sicherheit, Verträglichkeit und Wirksamkeit der Kombinationstherapie mit Venetoclax, Daratumumab und Dexamethason mit und ohne Bortezomib bei Teilnehmern mit rezidiviertem oder refraktärem multiplen Myelom zu bewerten.
Ziel:
In Teil 1 dieser Studie soll die objektive Ansprechrate in einem Zeitraum von bis zu 3 Monaten und die Anzahl der Teilnehmer mit dosisbegrenzenden Nebenwirkungen (Toxizitäten) ermittelt werden.
In Teil 2 dieser Studie soll die Anzahl der Teilnehmer mit dosisbegrenzenden Nebenwirkungen und das vollständige Ansprechen in einem Zeitraum von bis zu 5 Monaten ermittelt werden.
Hintergrund:
Venetoclax ist ein BCL-2-Inhibitor, der zur Behandlung der chronisch lymphatischen Leukämie bereits zugelassen ist. Es zählt chemisch gesehen zu den kleinen Molekülen, auch small molecules" genannt.
Venetoclax hemmt selektiv das Protein Bcl-2 das bei verschiedenen Krebsarten, einschließlich Multiples Myelom vermehrt vorkommt. Bcl-2 unterdrückt den programmierten Zelltod (Apoptose) der Tumorzellen. Venetoclax hingegen blockiert Bcl-2 im Zellinneren und führt so zum programmierten Zelltod der Tumorzellen. Dadurch wird auch die Wirksamkeit herkömmlicher Chemotherapeutika verbessert.
Einschlusskriterien:
- ECOG (Index zur Abstufung der Lebensqualität von Patienten mit Krebserkrankungen) von 0, 1 oder 2
- Rezidiviertes oder refraktäres Multiples Myelom mit dokumentierter Progression, die während oder nach dem letzten Behandlungsschema aufgetreten ist und vom Prüfer anhand der Kriterien der International Myeloma Working Group (IMWG) bestimmt wurde
- anhand, muss der Teilnehmer t(11;14) positiv sein [nachgewiesen in einem Zentrallabor durch einen sogenannten analytisch validierten Fluoreszenz-in-Situ-Hybridisierungs-(FISH)-Assay]
Ausschlusskriterien:
- Frühere Behandlung mit Venetoclax oder einem anderen B-Zell-Lymphoma-2-(BCL-2-) Inhibitor ODER frühere Behandlung mit Daratumumab oder einer anderen Anti-CD38-Therapie.
- Für Teilnehmer von Teil 2:
- Der Teilnehmer ist gegenüber einem Proteasom-Inhibitor refraktär, was definiert ist als Progression an oder innerhalb von 60 Tagen nach der letzten Dosis eines Behandlungsschemas mit einem Proteasominhibitor
- Behandlung mit einem Proteasom-Inhibitor innerhalb von 60 Tagen vor der ersten Dosis des Studienmedikaments
- Behandlung mit Antimyelom-Chemotherapie, Strahlentherapie, biologischer Therapie, Immuntherapie oder einer Forschungstherapie, einschließlich gezielter niedermolekularer Wirkstoffe innerhalb von 2 Wochen oder gegebenenfalls 5 Halbwertszeiten vor der ersten Dosis.
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt
Krefeld
Onkologische Gemeinschaftspraxis Dr. M. Neise und Dr. A. Lollert
02151-78025-0
Lemgo
Klinikum Lippe, Onkologie und Hämatologie
05261-26-0
Leverkusen
Klinikum Leverkusen, Medizinische Klinik III
0214-13-0
Mönchengladbach
Krankenhaus Maria Hilf GmbH, Franziskuskrankenhaus. Med. Klinik I
02161-892-2344
Studien
GMMG-HD8/DSMM-XIX
Induktionstherapie mit Lenalidomid, Bortezomib und Dexamethason und entweder intravenösem oder subkutanem Isatuximab bei Patienten mit neu diagnostiziertem Multiplem Myelom, die für eine Hochdosis-Chemotherapie mit nachfolgender autologer Stammzelltransplantation geeignet sind
Organisatorische Daten:
| Prüfplancode: | GMMG-HD 8 / DSMM XIX |
| ISRCTN: | |
| EudraCT: | 2022-000996-38 |
| Clinicaltrials.gov: | NCT05804032 |
| DRKS: | |
| Sponsor: | Universitätsklinikum Heidelberg DSMM, Sanofi und KKS-Netzwerk |
| Studienphase: | |
| Status: | Rekrutierung geplant bis Juni 2025 |
Ziel:
In dieser Studie für Patienten mit zuvor unbehandeltem Multiplem Myelom soll die Wirksamkeit der Induktionstherapie mit Isatuximab in Kombination mit Lenalidomid/Bortezomib/Dexamethason (RVd) bei Gabe von Isatuximab unter die Haut (subkutan) gegenüber einer Gabe in die Vene (intravenös) untersucht werden.
Durchführung:
Randomisierung: Die Patienten werden vor der Induktionstherapie nach dem Zufallsverfahren (randomisiert) einem von 2 Studienarmen (A oder B) zugeordnet.
- Patienten in Arm A erhalten 3 Zyklen des monoklonalen Antikörpers Isatuximab intravenös in Kombination mit dem RVd-Schema (Lenalidomid, Bortezomib, Dexamethason). Jeder Zyklus hat eine Dauer von 42 Tagen.
- Patienten in Arm B erhalten 3 Zyklen Isatuximab subkutan in Kombination mit RVd.
Nach der Induktionstherapie erhalten die Patienten eine Standard-Intensivierung (in der Regel eine Mobilisierungstherapie auf Cyclophosphamid-Basis, Stammzellenentnahme und hochdosiertes Melphalan, gefolgt von einer autologen Stammzelltransplantation
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- Bestätigte Diagnose eines neu diagnostizierten behandlungsbedürftigen Multiplen Myeloms gemäß den überarbeiteten Diagnosekriterien der International Myeloma Working Group (IMWG))
- Der Patient ist für eine Hochdosis-Chemotherapie und eine autologe Stammzelltransplantation geeignet
- Messbare Krankheitsaktivität (gemäß der Definition im Prüfplan)
- Alter 18 bis 70 Jahre bei Studieneinschluss
Ausschlußkriterien:
- Bekannte Überempfindlichkeit (oder Kontraindikation) gegen einen der Bestandteile der Studientherapie
- Systemische Amyloid-Leichtketten-Amyloidose (außer lokalisierte Amyloid-Leichtketten -Amyloidose, die auf die Haut oder das Knochenmark beschränkt ist)
- Plasmazell-Leukämie
- Vorherige Chemo- oder Strahlentherapie in den letzten 5 Jahren (außer lokaler Strahlentherapie bei lokalem Fortschreiten des Myeloms)
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Mühlheim an der Ruhr
Hämatologie Mülheim an der Ruhr
0208-76981
Zusätzliche Information
Dr. med. Jan Schröder,
Facharzt für Innere Medizin / Hämatologie und Internistische Onkologie
Münster
Gemeinschaftspraxis für Hämatologie und Onkologie
0251-620 08-0
Universitätsklinikum Münster, Medizinische Klinik und Poliklinik A
PD. Dr. C. Khandanpour, 0251-83-55555
Studien
ELIASVergleich Stammzelltransplantation mit konventioneller Therapie
Phase-II-Studie zum Vergleich einer Induktionstherapie mit Isatuximab in Kombination mit Bortezomid, Lenalidomid, und Dexamethason (I-VRD) gegenüber einer Induktionstherapie (I-VRD) mit anschließender Hochdosis-Chemotherapie, gefolgt von einer Erhaltungstherapie mit Isatuximab und Lenalidomid bei Patienten mit neu diagnostiziertem Multiplem Myelom
Organisatorische Daten:
| Prüfplancode: | UZL22_01 |
| ISRCTN: | |
| EudraCT: | 2022-500453-16-00 |
| Clinicaltrials.gov: | NCT05665140 |
| DRKS: | |
| Sponsor: | Universitätsklinikum Schleswig-Holstein, Campus Lübeck |
| Studienphase: | Phase 2 |
| Status: | Rekrutierung läuft, geplant bis August 2027 |
Ziele:
In dieser Studie wird geprüft, ob ein Schema mit einer Induktionstherapie mit Isatuximab, Bortezomib, Lenalidomid und Dexamethason (I-VRD) mit anschließender Hochdosis-Chemotherapie und autologer Stammzelltransplantation und Erhaltung einer alleinigen Induktionstherapie mit Stammzellmobilisierung und -apherese sowie Konsolidierung und Erhaltung überlegen ist.
Hintergrund:
In den aktuellen Leitlinien wird empfohlen, dass Patienten mit neu diagnostiziertem Multiplem Myelom, die für eine Transplantation infrage kommen, mehrere Zyklen einer Induktionstherapie erhalten, gefolgt von einer Hochdosis-Chemotherapie mit Melphalan und einer autologen Stammzelltransfusion.
Bei MM-Patienten kann hochdosiertes Melphalan das Gesamtüberleben und das progressionsfreie Überleben verbessern, doch kann diese Hochdosis-Chemotherapie zu akuten Toxizitäten und Langzeitfolgen oder in seltenen Fällen sogar zum Tode führen. Daher bleibt zu klären, ob eine hochdosierte Melphalan-Therapie auch Patienten mit niedrigem Risiko einen zusätzlichen Nutzen bietet oder ob diese Patienten mit einer intensivierten Induktionsbehandlung ohne anschließende hochdosierte Melphalan-Chemotherapie ausreichend behandelt werden können.
Durchführung:
Die Patienten werden nach dem Zufallsverfahren (randomisiert) einem von zwei Behandlungsarmen zugeordnet und erhalten entweder
- 3 Zyklen einer Induktionstherapie (I-VRD) + Stammzellmobilisierung und -apherese + 3 Zyklen einer Konsolidierung (I-VRD) (experimenteller Behandlungsarm)
oder - 3 Zyklen einer Induktionstherapie (I-VRD) + Stammzellmobilisierung und -apherese + Hochdosis-Chemotherapie mit Melphalan + autologe Stammzelltransplantation (Vergleichsarm)
In beiden Behandlungsgruppen folgt eine Erhaltung mit Isatuximab und Lenalidomid.
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- Neu diagnostiziertes, unbehandeltes, symptomatisches, dokumentiertes Myelom (gemäß der Definition im Prüfplan)
- Vorhandensein einer messbaren Erkrankung (gemäß der Definition im Prüfplan)
- Stadium I33 gemäß überarbeitetem Internationalen Stagingsystem (R-ISS)
Ausschlusskriterien:
- Hämolytische Anämie (mit Nachweis im direkten Coombs-Test)
- Beteiligung des zentralen Nervensystems (ZNS)
- Vorgeschichte oder Vorhandensein einer klinisch relevanten ZNS-Erkrankung wie Epilepsie, Krampfanfall, Parese, Aphasie, Schlaganfall, Subarachnoidalblutung oder andere ZNS-Blutung, schwere Hirnverletzungen, Demenz, Parkinson-Krankheit, Kleinhirnkrankheit, organisches Hirnsyndrom oder Psychose
- Patienten mit aktiver oder früherer Plasmazell-Leukämie, Waldenström-Makroglobulinämie, POEMS-Syndrom oder klinisch signifikanter Amyloidose
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt
Induktionstherapie mit Lenalidomid, Bortezomib und Dexamethason und entweder intravenösem oder subkutanem Isatuximab bei Patienten mit neu diagnostiziertem Multiplem Myelom, die für eine Hochdosis-Chemotherapie mit nachfolgender autologer Stammzelltransplantation geeignet sind
Organisatorische Daten:
| Prüfplancode: | GMMG-HD 8 / DSMM XIX |
| ISRCTN: | |
| EudraCT: | 2022-000996-38 |
| Clinicaltrials.gov: | NCT05804032 |
| DRKS: | |
| Sponsor: | Universitätsklinikum Heidelberg DSMM, Sanofi und KKS-Netzwerk |
| Studienphase: | |
| Status: | Rekrutierung geplant bis Juni 2025 |
Ziel:
In dieser Studie für Patienten mit zuvor unbehandeltem Multiplem Myelom soll die Wirksamkeit der Induktionstherapie mit Isatuximab in Kombination mit Lenalidomid/Bortezomib/Dexamethason (RVd) bei Gabe von Isatuximab unter die Haut (subkutan) gegenüber einer Gabe in die Vene (intravenös) untersucht werden.
Durchführung:
Randomisierung: Die Patienten werden vor der Induktionstherapie nach dem Zufallsverfahren (randomisiert) einem von 2 Studienarmen (A oder B) zugeordnet.
- Patienten in Arm A erhalten 3 Zyklen des monoklonalen Antikörpers Isatuximab intravenös in Kombination mit dem RVd-Schema (Lenalidomid, Bortezomib, Dexamethason). Jeder Zyklus hat eine Dauer von 42 Tagen.
- Patienten in Arm B erhalten 3 Zyklen Isatuximab subkutan in Kombination mit RVd.
Nach der Induktionstherapie erhalten die Patienten eine Standard-Intensivierung (in der Regel eine Mobilisierungstherapie auf Cyclophosphamid-Basis, Stammzellenentnahme und hochdosiertes Melphalan, gefolgt von einer autologen Stammzelltransplantation
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- Bestätigte Diagnose eines neu diagnostizierten behandlungsbedürftigen Multiplen Myeloms gemäß den überarbeiteten Diagnosekriterien der International Myeloma Working Group (IMWG))
- Der Patient ist für eine Hochdosis-Chemotherapie und eine autologe Stammzelltransplantation geeignet
- Messbare Krankheitsaktivität (gemäß der Definition im Prüfplan)
- Alter 18 bis 70 Jahre bei Studieneinschluss
Ausschlußkriterien:
- Bekannte Überempfindlichkeit (oder Kontraindikation) gegen einen der Bestandteile der Studientherapie
- Systemische Amyloid-Leichtketten-Amyloidose (außer lokalisierte Amyloid-Leichtketten -Amyloidose, die auf die Haut oder das Knochenmark beschränkt ist)
- Plasmazell-Leukämie
- Vorherige Chemo- oder Strahlentherapie in den letzten 5 Jahren (außer lokaler Strahlentherapie bei lokalem Fortschreiten des Myeloms)
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Neuss
Schwerpunktpraxis für Hämatologie und Onkologie
Dr. med. Christoph Losem, Dr. med. Dirk Plewe, 02131-101206
Olpe
Onkologische Schwerpunktpraxis, Medizinisches Versorgungszentrum 2 im Kreis Olpe
02761-926250
Paderborn
Brüderkrankenhaus St. Joseph
05251-702-0
Studien
GMMG-HD8/DSMM-XIX
Induktionstherapie mit Lenalidomid, Bortezomib und Dexamethason und entweder intravenösem oder subkutanem Isatuximab bei Patienten mit neu diagnostiziertem Multiplem Myelom, die für eine Hochdosis-Chemotherapie mit nachfolgender autologer Stammzelltransplantation geeignet sind
Organisatorische Daten:
| Prüfplancode: | GMMG-HD 8 / DSMM XIX |
| ISRCTN: | |
| EudraCT: | 2022-000996-38 |
| Clinicaltrials.gov: | NCT05804032 |
| DRKS: | |
| Sponsor: | Universitätsklinikum Heidelberg DSMM, Sanofi und KKS-Netzwerk |
| Studienphase: | |
| Status: | Rekrutierung geplant bis Juni 2025 |
Ziel:
In dieser Studie für Patienten mit zuvor unbehandeltem Multiplem Myelom soll die Wirksamkeit der Induktionstherapie mit Isatuximab in Kombination mit Lenalidomid/Bortezomib/Dexamethason (RVd) bei Gabe von Isatuximab unter die Haut (subkutan) gegenüber einer Gabe in die Vene (intravenös) untersucht werden.
Durchführung:
Randomisierung: Die Patienten werden vor der Induktionstherapie nach dem Zufallsverfahren (randomisiert) einem von 2 Studienarmen (A oder B) zugeordnet.
- Patienten in Arm A erhalten 3 Zyklen des monoklonalen Antikörpers Isatuximab intravenös in Kombination mit dem RVd-Schema (Lenalidomid, Bortezomib, Dexamethason). Jeder Zyklus hat eine Dauer von 42 Tagen.
- Patienten in Arm B erhalten 3 Zyklen Isatuximab subkutan in Kombination mit RVd.
Nach der Induktionstherapie erhalten die Patienten eine Standard-Intensivierung (in der Regel eine Mobilisierungstherapie auf Cyclophosphamid-Basis, Stammzellenentnahme und hochdosiertes Melphalan, gefolgt von einer autologen Stammzelltransplantation
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- Bestätigte Diagnose eines neu diagnostizierten behandlungsbedürftigen Multiplen Myeloms gemäß den überarbeiteten Diagnosekriterien der International Myeloma Working Group (IMWG))
- Der Patient ist für eine Hochdosis-Chemotherapie und eine autologe Stammzelltransplantation geeignet
- Messbare Krankheitsaktivität (gemäß der Definition im Prüfplan)
- Alter 18 bis 70 Jahre bei Studieneinschluss
Ausschlußkriterien:
- Bekannte Überempfindlichkeit (oder Kontraindikation) gegen einen der Bestandteile der Studientherapie
- Systemische Amyloid-Leichtketten-Amyloidose (außer lokalisierte Amyloid-Leichtketten -Amyloidose, die auf die Haut oder das Knochenmark beschränkt ist)
- Plasmazell-Leukämie
- Vorherige Chemo- oder Strahlentherapie in den letzten 5 Jahren (außer lokaler Strahlentherapie bei lokalem Fortschreiten des Myeloms)
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Siegburg
ZAHO -Rheinland
02241-59540
Studien
GMMG-HD8/DSMM-XIX
Induktionstherapie mit Lenalidomid, Bortezomib und Dexamethason und entweder intravenösem oder subkutanem Isatuximab bei Patienten mit neu diagnostiziertem Multiplem Myelom, die für eine Hochdosis-Chemotherapie mit nachfolgender autologer Stammzelltransplantation geeignet sind
Organisatorische Daten:
| Prüfplancode: | GMMG-HD 8 / DSMM XIX |
| ISRCTN: | |
| EudraCT: | 2022-000996-38 |
| Clinicaltrials.gov: | NCT05804032 |
| DRKS: | |
| Sponsor: | Universitätsklinikum Heidelberg DSMM, Sanofi und KKS-Netzwerk |
| Studienphase: | |
| Status: | Rekrutierung geplant bis Juni 2025 |
Ziel:
In dieser Studie für Patienten mit zuvor unbehandeltem Multiplem Myelom soll die Wirksamkeit der Induktionstherapie mit Isatuximab in Kombination mit Lenalidomid/Bortezomib/Dexamethason (RVd) bei Gabe von Isatuximab unter die Haut (subkutan) gegenüber einer Gabe in die Vene (intravenös) untersucht werden.
Durchführung:
Randomisierung: Die Patienten werden vor der Induktionstherapie nach dem Zufallsverfahren (randomisiert) einem von 2 Studienarmen (A oder B) zugeordnet.
- Patienten in Arm A erhalten 3 Zyklen des monoklonalen Antikörpers Isatuximab intravenös in Kombination mit dem RVd-Schema (Lenalidomid, Bortezomib, Dexamethason). Jeder Zyklus hat eine Dauer von 42 Tagen.
- Patienten in Arm B erhalten 3 Zyklen Isatuximab subkutan in Kombination mit RVd.
Nach der Induktionstherapie erhalten die Patienten eine Standard-Intensivierung (in der Regel eine Mobilisierungstherapie auf Cyclophosphamid-Basis, Stammzellenentnahme und hochdosiertes Melphalan, gefolgt von einer autologen Stammzelltransplantation
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- Bestätigte Diagnose eines neu diagnostizierten behandlungsbedürftigen Multiplen Myeloms gemäß den überarbeiteten Diagnosekriterien der International Myeloma Working Group (IMWG))
- Der Patient ist für eine Hochdosis-Chemotherapie und eine autologe Stammzelltransplantation geeignet
- Messbare Krankheitsaktivität (gemäß der Definition im Prüfplan)
- Alter 18 bis 70 Jahre bei Studieneinschluss
Ausschlußkriterien:
- Bekannte Überempfindlichkeit (oder Kontraindikation) gegen einen der Bestandteile der Studientherapie
- Systemische Amyloid-Leichtketten-Amyloidose (außer lokalisierte Amyloid-Leichtketten -Amyloidose, die auf die Haut oder das Knochenmark beschränkt ist)
- Plasmazell-Leukämie
- Vorherige Chemo- oder Strahlentherapie in den letzten 5 Jahren (außer lokaler Strahlentherapie bei lokalem Fortschreiten des Myeloms)
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Siegen
St.-Marien-Krankenhaus
0271 2310
Würselen
Hämatologisch-Onkologische Praxis, Dr. med. Christoph Maintz, Matthias Groschek, Dr. med. Christiane Hinske
02405–48920
Unterkategorien
-
Bonn
- COM_STUDIES_NUM
- 2
-
Dortmund
- COM_STUDIES_NUM
- 1
-
Würselen
- COM_STUDIES_NUM
- 1
-
Eschweiler
- COM_STUDIES_NUM
- 1
-
Bochum
- COM_STUDIES_NUM
- 2
-
Aachen
- COM_STUDIES_NUM
- 1
-
Duisburg
- COM_STUDIES_NUM
- 3
-
Bielefeld
- COM_STUDIES_NUM
- 3
-
Essen
- COM_STUDIES_NUM
- 3
-
Düsseldorf
- COM_STUDIES_NUM
- 2
-
Hagen
- COM_STUDIES_NUM
- 1
-
Hamm
- COM_STUDIES_NUM
- 1
-
Köln
- COM_STUDIES_NUM
- 2
-
Krefeld
- COM_STUDIES_NUM
- 1
-
Lemgo
- COM_STUDIES_NUM
- 1
-
Leverkusen
- COM_STUDIES_NUM
- 1
-
Mönchengladbach
- COM_STUDIES_NUM
- 1
-
Mühlheim an der Ruhr
- COM_STUDIES_NUM
- 1
-
Münster
- COM_STUDIES_NUM
- 2
-
Neuss
- COM_STUDIES_NUM
- 1
-
Olpe
- COM_STUDIES_NUM
- 1
-
Siegburg
- COM_STUDIES_NUM
- 1
-
Goch
- COM_STUDIES_NUM
- 1
-
Siegen
- COM_STUDIES_NUM
- 1
-
Paderborn
- COM_STUDIES_NUM
- 1
