COM_STUDIES_DEFAULT_PAGE_TITLE
Köln
Köln
Onkologische Gemeinschaftspraxis Köln
0221 - 931 822 0
Universitätsklinikum Köln Medizinische Klinik I
0221-478-96548
Studien
DREAMM 7Belantamab-Mafodotin
Bewertung der Wirksamkeit und Sicherheit von Belantamab-Mafodotin, Bortezomib und Dexamethason gegenüber Daratumumab, Bortezomib und Dexamethason bei Teilnehmern mit rezidiviertem/refraktärem Multiplen Myelom
Organisatorische Daten:
| Prüfplancode: | 207503 |
| ISRCTN: | |
| EudraCT: | 2018-003993-29 |
| Clinicaltrials.gov: | NCT04246047 |
| DRKS: | |
| Sponsor: | GlaxoSmithKline |
| Studienphase: | III |
| Status: | Rekrutierung geplant bis Juni/2026 |
Kurzbeschreibung:
Hierbei handelt es sich um eine randomisierte, offene Phase-3-Studie zur Untersuchung der Sicherheit und Wirksamkeit von Belantamab-Mafodotin in Kombination mit Bortezomib/Dexamethason (Arm A) gegenüber Daratumumab in Kombination mit Bortezomib/Dexamethason (Arm B) bei Teilnehmern mit rezidivierendem multiplem Myelom..
Hintergrund:
Die amerikanische Arzneimittelbehörde FDA hat Belantamab-Mafodotin-blmf (GSK2857916; Blenrep) für die Behandlung von Patienten mit rezidiviertem oder refraktärem Multiplen Myelom zugelassen, die zuvor mit mindestens 4 Therapien behandelt wurden, darunter ein immunmodulatorisches Mittel, ein Proteasom-Inhibitor und ein Anti-CD38-Antikörper. Dies ist weltweit die erste Anti-BCMA-Behandlung, die bei dieser Patientenpopulation verfügbar ist.
Belantamab-Mafodotin ist gegen das B-Zell-Reifungsantigen (BCMA) gerichtet, das eine wichtige Rolle für das Überleben von Plasmazellen spielt und auf Zellen des Multiplen Myeloms exprimiert wird. Das Antikörper-Wirkstoff-Konjugat Belantamab-Mafodotin bindet an BCMA auf Myelom-Zelloberflächen, wodurch der Zellzyklus angehalten und eine antikörperabhängige zelluläre Zytotoxizität induziert wird.
Normalerweise fördert BCMA das Überleben von Plasmazellen durch Signalübertragung von zwei bekannten Liganden. Dieser Signalweg hat sich als wichtig für das Wachstum und Überleben von Myelomzellen erwiesen.
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- Bestätigte Diagnose eines Multiplen Myeloms (MM) gemäß IMWG-Kriterien
- Vorbehandlung des MM mit mindestens einer Therapielinie und dokumentierte Krankheitsprogression während oder nach der letzten Therapie.
- ECOG Performance Status (Index zur Abstufung der Lebensqualität von Patienten mit Krebserkrankungen) von 0 bis 2
- Muss mindestens 1 Aspekt einer messbaren Erkrankung aufweisen, wie folgt definiert:
- Urin-M-Protein-Ausscheidung > 200 mg pro 24 Stunden, oder
- Serum-M-Protein-Konzentration > 0,5 g/dl, oder
- Freie Leichtketten im Serum (FLC): beteiligter FLC-Spiegel > 10 mg/dl (> 100 mg/L) und ein abnormes Verhältnis der freien Leichtketten im Serum (< 0,26 oder > 1,65).
- Alle Nebenwirkungen, die infolge durch Vorbehandlungen hervorgerufen wurden, müssen zum Zeitpunkt der Rekrutierung < Grad 1 sein, mit Ausnahme von Alopezie.
Ausschlußkriterien:
- Unverträglichkeit gegenüber Daratumumab.
- Kein bzw. kein hinreichendes Ansprechen (therapierefraktär) gegenüber Daratumumab oder einer anderen Anti-CD38-Therapie (definiert als fortschreitende Erkrankung während der Behandlung mit einer Anti-CD38-Therapie oder innerhalb von 60 Tagen nach Abschluss dieser Behandlung).
- Unverträglichkeit gegenüber Bortezomib oder kein bzw. kein hinreichendes Ansprechen (therapierefraktär) gegenüber Bortezomib (definiert als fortschreitende Erkrankung während der Behandlung mit einem Bortezomib-haltigen Schema von 1,3 mg/m2 zweimal wöchentlich oder innerhalb von 60 Tagen nach Abschluss dieser Behandlung). Hinweis: Patienten mit fortschreitender Erkrankung während der Behandlung mit einem wöchentlichen Bortezomib-Schema dürfen teilnehmen.
- Anhaltende periphere Neuropathie von Grad 2 oder höher oder neuropathische Schmerzen.
- Vorherige Behandlung mit einer Anti-B-Zellreifungsantigen- (Anti-BCMA-) Therapie.
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Daratumumab zur Erstlinienbehandlung von Patienten mit Myelom, die für eine Transplantation nicht in Frage kommen, gefolgt von einer erneuten Daratumumab-Behandlung beim ersten Rückfall (Rezidiv)
.Organisatorische Daten:
| Prüfplancode: | Uni-Koeln-3946 |
| ISRCTN: | |
| EudraCT: | 2019-003856-35 |
| Clinicaltrials.gov: | NCT04656951 |
| DRKS: | |
| Sponsor: | Universitätsklinnikum Köln/Janssen |
| Studienphase: | Phase 2 |
| Status: | Rekrutierung läuft , bis Dez 2029 |
Kurzbeschreibung
Dies ist eine Studie zur Bewertung der Wirksamkeit und Sicherheit des Medikaments Daratumab bei Patienten mit unbehandeltem Multiplem Myelom (MM).
Untersucht wird:
1. die Sicherheit und Wirksamkeit von Daratumumab als Zusatz zu einem Standardinduktionsregimes mit Bortezomib, Cyclophosphamid und Dexamethason (VCD)
2. die Wirksamkeit der Erhaltungstherapie bis zur Progression unter Anwendung von Daratumumab in Kombination mit Bortezomib und Dexamethason
3. die Wirksamkeit eines Daratumumab-haltigen Regimes beim ersten Rezidiv nach einem Daratumumab-haltigen Erstlinienregime
Hintergrund:
Daratumumab ist ein monoklonaler Antikörper, der zur Behandlung des Multiplen Myeloms eingesetzt wird. Der Antikörper bindet an das Oberflächenprotein CD38, das von den Tumorzellen in allen Stadien der Erkrankung gebildet wird. Die Bindung des Antikörpers führt zum Absterben der Tumorzelle durch Apoptose sowie durch eine verstärkte Immunreaktion.
VCD (Velcade, Cyclophosphamid, Dexamethason) hat sich in Deutschland als Standard für die Erstlinientherapie von Patienten etabliert, die für eine Transplantation in Frage kommen. Aufgrund des günstigen Nebenwirkungsprofils ist dieses Regime auch für Patienten geeignet, die für eine Transplantation nicht in Frage kommen. Daratumumab (Dara) hat sich in einer Vielzahl von Kombinationen wie Bortezomib/Melphalan/Prednison (VMP), Bortezomib/Dexamethason (Vd) oder Lenalidomid/Dexamethason (Rd) als sicher und wirksam erwiesen. Unter Verwendung einer reduzierten Cyclophosphamid-Dosis soll in der Studie die Sicherheit und Wirksamkeit von Dara in Kombination mit VCD (Dara-VCD) bei Patienten untersucht werden, die für eine Transplantation nicht in Frage kommen.
Die Erhaltungstherapie ist bei Patienten, die für eine Transplantation in Frage kommen, und bei Patienten, die nicht dafür in Frage kommen, zum Standard geworden. Es besteht jedoch Unsicherheit darüber, ob eine Erhaltungstherapie mit einem einzigen Wirkstoff ausreicht oder ob eine Kombinationstherapie erforderlich ist. In dieser Studie wird die Sicherheit und Wirksamkeit der Erhaltungstherapie mit Daratumumab in Kombination mit Bortezomib und Dexamethason untersucht.
Die Vielzahl der Behandlungsmöglichkeiten beim ersten Rückfall hat dazu geführt, dass eine optimale Abfolge von Therapieschemata in der Erst- und Zweitlinie gefunden werden muss. Insbesondere ist unklar, ob die Wirksamkeit von Daratumumab bei einem Rückfall beeinflusst wird, wenn es vorher als Teil der Erstlinienbehandlung verwendet wurde.
Einschlusskriterien:
- Patienten müssen ein unbehandeltes, dokumentiertes, behandlungsbedürftiges multiples Myelom haben, das folgenden Kriterien erfüllt:
- Monoklonale Plasmazellen im Knochenmark ≥ 10% und/oder Nachweis eines Plasmozytoms, nachgewiesen durch eine Biopsie
- Multiples Myelom IgG: Serumspiegel des monoklonalem Paraproteins (M-Protein) ≥ 1,0 g/dl oder M-Proteinspiegel im Urin ≥ 200 mg/24 Stunden; oder
- Multiples Myelom mit IgA, IgD, IgE, IgM: M-Proteinspiegel im Serum ≥ 0,5 g/dl oder M-Proteinspiegel im Urin ≥ 200 mg/24 Stunden; oder
- Multiples Leichtketten-Myelom ohne messbare Erkrankung im Serum oder Urin: Freie Leichtketten-Immunoglobuline im Serum ≥ 10 mg/dl und abnormales Verhältnis der freien Leichtketten-Immunoglobuline Kappa Lambda.
- ECOG < 2 (Index zur Abstufung der Lebensqualität von Patienten mit Krebserkrankungen)
Ausschlusskriterien:
- Vorherige Therapie des multiplen Myeloms, mit Ausnahme von Dexamethason in einer Höchstdosis von insgesamt 160 mg, einer Notfall-Strahlentherapie oder einer Operation zur Linderung der Symptome
- Teilnahme an anderen interventionellen klinischen Studien
- bekannte meningeale Beteiligung des Multiplen Myeloms.
- Vorgeschichte mit einer bösartigen Erkrankung (außer dem Multiplen Myelom) innerhalb von 3 Jahren vor dem Datum des Screenings.
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt
CAR T-Zelltherapie mit bb2121
Eine Phase-2-Studie zur Bestimmung der Wirksamkeit und Sicherheit von bb2121 bei Patienten mit rezidiviertem (wieder aufgetretenem) und refraktärem (nicht auf Behandlung ansprechendem) Multiplem Myelom (RRMM) und bei Patienten mit risikoreichem Multiplem Myelom, bei denen die Krankheit innerhalb von 18 Monaten nach der Erstbehandlung fortgeschritten ist.
Organisatorische Daten:
| Prüfplancode: | KarMMa2 |
| ISRCTN: | |
| EudraCT: | 2018-000264-28 |
| Clinicaltrials.gov: | NCT03601078 |
| DRKS: | |
| Sponsor: | Celgene |
| Studienphase: | Phase 2 |
| Status: | Rekrutierung läuft, geplant bis März 2025 |
Ziel:
In dieser Studie soll die Wirksamkeit und Sicherheit von bb2121 bei Patienten mit rezidiviertem oder refraktären Multiplen Myelom bewertet werden.
Hintergrund:
Die chimäre-Antigenrezeptor-(CAR) T-Zelltherapie bb2121 ist gegen das Oberflächenantigen BCMA (engl. B-cell maturation antigen) gerichtet, um T-Zellen dahin gehend zu programmieren, dass sie maligne Myelomzellen erkennen und abtöten. https://meetinglibrary.asco.org/record/160693/abstract
In einer Phase-1-Studie bei stark vorbehandelten Patienten mit rezidiviertem oder refraktären multiplem Myelom zeigte bb2121 ermutigende Ergebnisse mit tiefem und dauerhaftem Ansprechen bei gleichzeitig guter Verträglichkeit. https://www.presseportal.de/pm/62626/3972298 In der vorliegenden Studie soll die Wirksamkeit und Sicherheit von bb2121 als mögliche Therapie für Multiples Myelom weiter untersucht werden.
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- Es besteht eine messbare Krankheit, definiert als
- M-Protein (Serum-Protein-Elektrophorese [sPEP] oder Urin-Protein-Elektrophorese [uPEP]): sPEP ≥ 0,5 g/dl oder uPEP ≥ 200 mg/24 Stunden und/oder
- Leichtketten-MM ohne messbare Krankheit im Serum oder Urin: freie Leichtketten im Serum ≥ 10 mg/dl und abnormes Kappa-Lambda-Leichtketten-Verhältnis im Serum
- Spezifische Anforderungen für:
- Kohorte 1: ≥ 3 frühere Anti-Myelom-Behandlungsschemen
- Kohorte 2: 1 frühere Anti-Myelom-Behandlung
- ECOG Performance Status von 0 oder 1 ((Index zur Abstufung der Lebensqualität von Patienten mit Krebserkrankungen)
Ausschlusskriterien:
- Erhalt eines Prüfpräparats innerhalb von 14 Tagen vor der Leukapherese
- Eines der folgenden Verfahren innerhalb der letzten 14 Tage vor der Leukapherese:
- Plasmapherese
- Große Operation (nach Definition des Prüfarztes)
- Strahlentherapie mit Ausnahme der lokalen Therapie für myelombedingten Knochenläsionen
- Anwendung einer systemischen Anti-Myelom-Medikamententherapie
- Bekannte Beteiligung des zentralen Nervensystems
- Klinischer Nachweis einer Lungenleukostase und Verbrauchskoagulopathie (disseminierten intravasale Gerinnung)
- Vorgeschichte oder Vorliegen einer klinisch relevanten Erkrankung des Zentralnervensystems (ZNS)
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Ergänzende Informationen
Zur Herstellung der mit einem bb2121 chimären Antigenrezeptor (CAR) modifizierten T-Zellen werden die weißen Blutkörperchen (Leukozyten) aus dem Blut herausgelöst. Dies bezeichnet man als Leukapherese. Vor der bb2121-Infusion wird bei den Patienten ein Verfahren zur Entfernung der Lymphozyten, die sogenannte Lymphozytendepletion, mit Fludarabin und Cyclophosphamid durchgeführt.
CAR T-Zelltherapie mit bb2121
Es handelt sich um eine multizentrische, randomisierte, offene Phase-3-Studie, in der die Wirksamkeit und Sicherheit von bb2121 im Vergleich zu den Standardschemata bei Patienten mit rezidiviertem und refraktärem multiplem Myelom (RRMM) verglichen wird.
Organisatorische Daten:
| Prüfplancode: | KarMMa-3 |
| ISRCTN: | U1111-1217-9988 |
| EudraCT: | 2018-001023-38 |
| Clinicaltrials.gov: | NCT03651128 |
| DRKS: | |
| Sponsor: | Celgene |
| Studienphase: | Phase 3 |
| Status: | Rekrutierung läuft, geplant bis Juni 2025 |
Ziel:
In dieser Studie soll die Wirksamkeit und Sicherheit von bb2121 bei Patienten mit rezidiviertem oder refraktären Multiplen Myelom im Vergleich zu einer Standardtherapie bewertet werden.
Hintergrund:
Die chimäre-Antigenrezeptor-(CAR) T-Zelltherapie bb2121 ist gegen das Oberflächenantigen BCMA (engl. B-cell maturation antigen) gerichtet, um T-Zellen dahin gehend zu programmieren, dass sie maligne Myelomzellen erkennen und abtöten. https://meetinglibrary.asco.org/record/160693/abstract
In einer Phase-1-Studie bei stark vorbehandelten Patienten mit rezidiviertem oder refraktärem Multiplem Myelom zeigte bb2121 ermutigende Ergebnisse mit tiefem und dauerhaftem Ansprechen bei gleichzeitig guter Verträglichkeit. https://www.presseportal.de/pm/62626/3972298
In der vorliegenden Studie soll die Wirksamkeit und Sicherheit von bb2121 als mögliche Therapie für Multiples Myelom im Vergleich zu einer Standardbehandlung weiter untersucht werden.
Durchführung
 Es wird erwartet, dass in der Studie etwa 381 Patienten mit RRMM randomisiert werden. Etwa 254 Patienten werden in Behandlungsarm A und etwa 127 Patienten in Behandlungsarm B randomisiert.
Es wird erwartet, dass in der Studie etwa 381 Patienten mit RRMM randomisiert werden. Etwa 254 Patienten werden in Behandlungsarm A und etwa 127 Patienten in Behandlungsarm B randomisiert.
Zur Herstellung der mit einem bb2121 chimären Antigenrezeptor (CAR) modifizierten T-Zellen werden die weißen Blutkörperchen (Leukozyten) aus dem Blut herausgelöst. Dies bezeichnet man als Leukapherese. Die Leukozyten werden dann behandelt und zurückinfundiert. Vor der bb2121-Infusion wird bei den Patienten ein Verfahren zur Entfernung der Lymphozyten, die sogenannte Lymphozytendepletion, mit Fludarabin und Cyclophosphamid durchgeführt.
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- Patient hat eine messbare Krankheit, definiert als:
- M-Protein (Serum-Protein-Elektrophorese [sPEP] oder Urin-Protein-Elektrophorese [uPEP]): sPEP ≥ 0,5 g/dl oder uPEP ≥ 200 mg/24 Stunden und/oderLeichtketten-MM ohne messbare Krankheit im Serum oder Urin: immunglobulinfreie Leichtketten im Serum ≥ 10 mg/dl und abnormes Kappa-Lambda-Leichtketten-Verhältnis im Serum
- Patient hat mindestens 2, aber höchstens 4 vorherige Behandlungsschemen für MM erhalten.
- Patient hat mindestens 2 Behandlungszyklen mit DARA, einem Proteasom-Inhibitor- und einem immunmodulatorischen Wirkstoffschema für mindestens 2 aufeinanderfolgende Zyklen erhalten.
- Patient muss gegenüber dem letzten Behandlungsschema refraktär sein, also ein dokumentiertes Fortschreiten der Erkrankung während oder innerhalb von 60 Tagen nach Abschluss des letzten Anti-Myelom-Schemas vor Studieneinschluss aufweisen.
- ECOG Performance Status von 0 oder 1 (Index zur Abstufung der Lebensqualität von Patienten mit Krebserkrankungen
Ausschlusskriterien:
- Nichtsekretorisches multiples Myelom (MM)
- Bestimmte auffällige Laborwerte
- unzureichende Lungenfunktion, definiert als eine Sauerstoffsättigung von weniger als 92 % bei Raumluft
- Maligne Erkrankungen neben MM in der Vorgeschichte. Dieses Kriterium gilt nicht, wenn diese Erkrankungen nicht in den letzten 5 Jahren oder länger vorlagen
- Bekannte Beteiligung des zentralen Nervensystems
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Studie mit Elranatamab im Vergleich zu Lenalidomid bei Patienten mit neu diagnostiziertem Multiplem Myelom nach Transplantation
Organisatorische Daten:
| Prüfplancode: | C1071005, MagnetisMM-7 |
| ISRCTN: | |
| EudraCT: | 2021-006052-14 |
| Clinicaltrials.gov: | NCT05317416 |
| DRKS: | |
| Sponsor: | Pfizer |
| Studienphase: | Phase 3 |
| Status: | Rekrutierung läuft, geplant bis Juni 2027 |
Randomisierte, 2-armige Phase-3-Studie mit Elranatamab (PF-06863135) im Vergleich zu Lenalidomid bei Patienten mit neu diagnostiziertem Multiplem Myelom, die nach einer autologen Stammzelltransplantation eine minimale Resterkrankung aufweisen.
Ziel:
Mit dieser Studie soll untersucht werden, ob eine Elranatamab-Monotherapie im Vergleich zu einer Lenalidomid-Monotherapie Teilnehmern mit neu diagnostiziertem Multiplem Myelom einen klinischen Nutzen bietet, bei denen nach einer autologen Stammzelltransplantation noch eine minimale Resterkrankung nachweisbar ist.
Hintergrund:
Der bispezifische BCMA/CD3-Antikörper Elranatamab (PF-06863135) zeigte bei Patienten mit rezidiviertem/refraktärem Multiplem Myelom eine vielversprechende vorläufige Wirksamkeit und Verträglichkeit.
Daten aus der Phase-1-Studie MagnetisMM-1 (NCT03269136), die auf der ASCO-Jahrestagung 2021 vorgestellt wurden, zeigten, dass Elranatamab in Dosen von bis zu 1000 µg/kg ein überschaubares Sicherheitsprofil ohne dosislimitierende Toxizitäten aufweist.
Darüber hinaus erzielte der Wirkstoff bei Dosierungen von 215 µg/kg und höher eine Gesamtansprechrate (ORR) von 70 % und eine vollständige Ansprechrate (CR)/stringente CR von 30 %. Bei der empfohlenen Phase-2-Dosis von 1000 µg/kg erzielte der Wirkstoff eine ORR von 83,3 %. Bemerkenswert ist, dass drei von vier in die Studie aufgenommenen Patienten, die zuvor mit einer BCMA-gerichteten Therapie behandelt worden waren, ein Ansprechen erzielten.
Durchführung:
Die Teilnehmer an der Studie werden nach dem Zufallsprinzip einer von zwei Behandlungsgruppen zuteilt.
- Teilnehmer in Gruppe 1 erhalten Elrantamab als Injektion unter die Haut in der Studienklinik. Nach den anfangs an den gegenwärtigen Zustand des Patienten angepassten Dosen (sogenannte Step-up-Dosen) erhalten die Teilnehmer dann eine Dosis Elrantamab pro Woche. Nach 6 Monaten wird die Häufigkeit der Klinikbesuche für die Verabreichung der Injektionen auf jede zweite Woche reduziert.
- Teilnehmer in Gruppe 2 erhalten Lenalidomid oral einmal täglich zu Hause.
Die Teilnahme an der Studie dauert etwa fünf Jahre.
Ein und Ausschlusskriterien:
Einschlusskriterien:
-
Diagnose eines Multiplen Myeloms gemäß den von der International Myeloma Working Group (IMWG) aufgestellten Diagnosekriterien
-
Vorgeschichte einer Induktionstherapie für neu diagnostiziertes MM, gefolgt von einer Hochdosistherapie und einer autologen Stammzelltransplantation (ASZT). Die Randomisierung muss innerhalb von 120 Tagen nach der ASZT erfolgen. Bei Teilnehmern, die nach der ASZT eine Konsolidierungstherapie erhalten, muss die Randomisierung innerhalb von 60 Tagen nach der Konsolidierung und innerhalb von 6 Monaten nach der ASZT erfolgen.
-
Partielles Ansprechen oder besser nach den IMWG-Kriterien zum Zeitpunkt der RandomisierungNachweis einer minimalen Resterkrankung (MRD-positiv, ≥ 10-5) beim Screening.
-
ECOG Performance Status (Index zur Abstufung der Lebensqualität von Patienten mit Krebserkrankungen) von 0 oder 1.
Ausschlusskriterien:
-
Plasmazell-Leukämie
-
POEMS-Syndrom
-
Amyloidose, Waldenström-Makrogobulinämie
-
Bekannte aktive Beteiligung des ZNS oder klinische Anzeichen einer Beteiligung der Hirnhäute (Meningen)
-
Vorherige MM-Erhaltungstherapie
-
Vorherige Behandlung mit einer BCMA-Therapie
-
Jede andere aktive bösartige Erkrankung innerhalb von 3 Jahren vor der Aufnahme in die Studie, mit Ausnahme von adäquat behandeltem Basalzell- oder Plattenepithelkarzinom der Haut oder Karzinom in situ (z. B. bei der Diagnose) oder vor der Transplantation.
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt
Talquetamab
Talquetamab in Kombination mit Daratumumab oder in Kombination mit Daratumumab und Pomalidomid im Vergleich zu Daratumumab in Kombination mit Pomalidomid und Dexamethason bei Teilnehmern mit rezidiviertem oder refraktärem Multiplem Myelom (MonumenTAL-3)
Organisatorische Daten:
| Prüfplancode: | CR109082, 64407564MMY3002 |
| ISRCTN: | |
| EudraCT: | 2021-000202-22 |
| Clinicaltrials.gov: | NCT05455320 |
| DRKS: | |
| Sponsor: | Janssen |
| Studienphase: | 3 |
| Status: | Rekrutierung geplant bis Febr 2029 |
Ziel:
Ziel der Studie ist der Vergleich der Wirksamkeit von unter die Haut (subkutan, s.c.) verabreichtem Talquetamab in Kombination mit Daratumumab s.c. und Pomalidomid (Tal-DP) bzw. Talquetamab s.c. in Kombination mit Daratumumab s.c. (Tal-D) gegenüber Daratumumab s.c. in Kombination mit Pomalidomid und Dexamethason (DPd) bei Teilnehmern mit rezidiviertem oder refraktärem Multiplem Myelom, die mindestens eine vorherige Therapielinie erhalten haben.
Hintergrund:
Talquetamab ist ein humanisierter monoklonaler Antikörper, der gegen zwei Ziele gerichtet (bispezifisch) ist: gegen humanes CD3, ein Oberflächenantigen auf T-Zellen, und gegen GPRC5D (G protein-coupled receptor family C group 5 member D), ein Antigen in Tumorzellen oder auf deren Oberfläche (tumor-assoziiertes Antigen). Bei Verabreichung bindet Talquetamab sowohl an CD3 auf T-Zellen als auch an GPRC5D, das auf bestimmten Tumorzellen wie z.B. dem Multiplen Myelom vermehrt vorkommt und eine Schlüsselrolle bei der Vermehrung von Tumorzellen spielt.
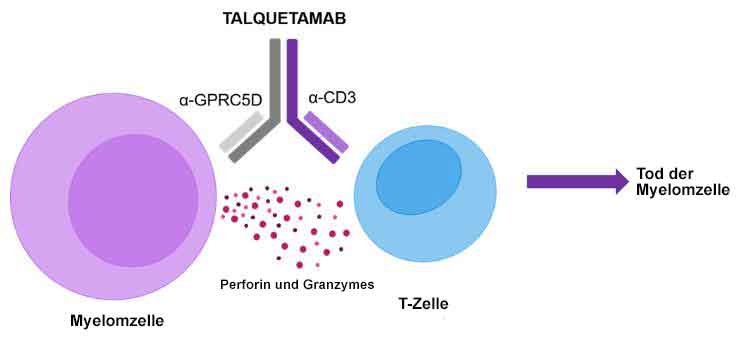
Abb. aus https://multiplemyelomahub.com/,
Durchführung:
Die Studie ist in drei Phasen unterteilt: Voruntersuchung (Screening), Behandlung (bis eines der folgenden Ereignisse eintritt: bestätigtes Fortschreiten der Krankheit, Tod, unverträgliche Toxizität, Widerruf der Einwilligung oder Ende der Studie) und Nachbeobachtung (bis eines der folgenden Ereignisse eintritt: Tod, Widerruf der Einwilligung, Verlust der Nachbeobachtung oder Ende der Studie). Wirksamkeit, Sicherheit, Pharmakokinetik, Immunogenität und Biomarker werden zu bestimmten Zeitpunkten untersucht. Die Gesamtdauer der Studie beträgt bis zu 6 Jahren und 6 Monaten.
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- Dokumentiertes Multiples Myelom, definiert als
- a) Diagnose des Multiplen Myeloms gemäß den diagnostischen Kriterien der International Myeloma Working Group (IMWG) und
- b) messbare Erkrankung zum Zeitpunkt des Screenings gemäß der Definition im Protokoll
- Rezidivierte oder refraktäre Erkrankung gemäß folgender Definition:
- eine rezidivierte Erkrankung ist definiert als anfängliches Ansprechen auf die vorherige Behandlung, gefolgt von einer nach IMWG-Kriterien bestätigten fortschreitenden (progredienten) Erkrankung mehr als 60 Tage nach Beendigung der Behandlung;
- eine refraktäre Erkrankung ist definiert als eine Verringerung des monoklonalen Paraproteins (M-Protein) um weniger als 25 % oder eine nach IMWG-Kriterien bestätigte fortschreitende Erkrankung während der vorherigen Behandlung oder ≤ 60 Tage nach Beendigung der Behandlung
- Teilnehmer müssen mindestens eine vorherige Linie einer Myelom-Therapie, einschließlich eines Proteasom-Inhibitors (PI) und Lenalidomid, erhalten haben. Patienten, die nur eine vorherige Linie einer Myelom-Therapie erhalten haben, müssen als Lenalidomid-refraktär gelten (d. h. bei ihnen ist bei Beendigung einer Lenalidomid-haltigen Therapie oder innerhalb von 60 Tagen danach eine fortschreitende (progrediente) Erkrankung nach IMWG-Kriterien nachweisbar). Teilnehmer, die 2 oder mehr vorherige Therapielinien zur Behandlung des Myeloms erhalten haben, müssen als Lenalidomid-behandelt gelten
- Dokumentierte Anzeichen einer fortschreitenden Erkrankung, basierend auf der Feststellung des Ansprechens durch den Prüfarzt gemäß den IMWG-Kriterien während oder nach der letzten Behandlung
- ECOG (Index zur Abstufung der Lebensqualität von Patienten mit Krebserkrankungen) von 0, 1 oder 2
Ausschlußkriterien
- Gegenanzeigen oder lebensbedrohliche Allergien, Überempfindlichkeiten oder Unverträglichkeiten gegenüber Hilfsstoffen des Studienmedikaments
- Refraktär gegenüber einem monoklonalen Antikörper gegen Cluster of Differentiation 38 (CD38), wie in den IMWG-Konsensrichtlinien definiert (Fortschreiten der Erkrankung während der Behandlung oder innerhalb von 60 Tagen nach Beendigung der Therapie mit einem monoklonalen Antikörper gegen CD38)
- Erhalt von Kortikosteroiden in einer maximalen Gesamtdosis von oder entsprechend ≥ 140 mg Prednison innerhalb von 14 Tagen vor Gabe der ersten Dosis des Studienmedikaments
- Bekannte aktive Beteiligung des zentralen Nervensystems (ZNS) oder klinische Anzeichen einer meningealen Beteiligung des Multiplen Myeloms. Bei Verdacht auf eine der beiden Erkrankungen sind eine negative Magnetresonanztomographie (MRT) des gesamten Gehirns und eine lumbale Zytologie erforderlich.
- Plasmazellleukämie zum Zeitpunkt der Voruntersuchung, Waldenström-Makroglobulinämie, Polyneuropathie, Organomegalie, Endokrinopathie, M-Protein und Hautveränderungen (POEMS-Syndrom) oder primäre Amyloid-Leichtketten-Amyloidose
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Venetoclax in Kombination mit Daratumumab /Dexamethason
Eine Studie zur Kombinationstherapie mit Venetoclax, Daratumumab und Dexamethason (mit und ohne Bortezomib) bei Patienten mit rezidiviertem oder refraktärem Multiplem Myelom.
Organisatorische Daten:
| Prüfplancode: | M15-654 |
| ISRCTN: | |
| EudraCT: | 2017-002099-26 |
| Clinicaltrials.gov: | NCT03314181 |
| DRKS: | |
| Sponsor: | AbbVie in Zusammenarbeit mit Janssen Research & Development, LLC |
| Studienphase: | Phase 1/2 |
| Status: | Rekrutierung läuft, geplant bis 6/2024 |
Kurzbeschreibung:
Multizentrische Dosissteigerungs- und Erweiterungsstudie der Phase 1 / 2 zur Bewertung der Sicherheit, Verträglichkeit und Wirksamkeit der Kombinationstherapie mit Venetoclax, Daratumumab und Dexamethason mit und ohne Bortezomib bei Teilnehmern mit rezidiviertem oder refraktärem multiplen Myelom zu bewerten.
Ziel:
In Teil 1 dieser Studie soll die objektive Ansprechrate in einem Zeitraum von bis zu 3 Monaten und die Anzahl der Teilnehmer mit dosisbegrenzenden Nebenwirkungen (Toxizitäten) ermittelt werden.
In Teil 2 dieser Studie soll die Anzahl der Teilnehmer mit dosisbegrenzenden Nebenwirkungen und das vollständige Ansprechen in einem Zeitraum von bis zu 5 Monaten ermittelt werden.
Hintergrund:
Venetoclax ist ein BCL-2-Inhibitor, der zur Behandlung der chronisch lymphatischen Leukämie bereits zugelassen ist. Es zählt chemisch gesehen zu den kleinen Molekülen, auch small molecules" genannt.
Venetoclax hemmt selektiv das Protein Bcl-2 das bei verschiedenen Krebsarten, einschließlich Multiples Myelom vermehrt vorkommt. Bcl-2 unterdrückt den programmierten Zelltod (Apoptose) der Tumorzellen. Venetoclax hingegen blockiert Bcl-2 im Zellinneren und führt so zum programmierten Zelltod der Tumorzellen. Dadurch wird auch die Wirksamkeit herkömmlicher Chemotherapeutika verbessert.
Einschlusskriterien:
- ECOG (Index zur Abstufung der Lebensqualität von Patienten mit Krebserkrankungen) von 0, 1 oder 2
- Rezidiviertes oder refraktäres Multiples Myelom mit dokumentierter Progression, die während oder nach dem letzten Behandlungsschema aufgetreten ist und vom Prüfer anhand der Kriterien der International Myeloma Working Group (IMWG) bestimmt wurde
- anhand, muss der Teilnehmer t(11;14) positiv sein [nachgewiesen in einem Zentrallabor durch einen sogenannten analytisch validierten Fluoreszenz-in-Situ-Hybridisierungs-(FISH)-Assay]
Ausschlusskriterien:
- Frühere Behandlung mit Venetoclax oder einem anderen B-Zell-Lymphoma-2-(BCL-2-) Inhibitor ODER frühere Behandlung mit Daratumumab oder einer anderen Anti-CD38-Therapie.
- Für Teilnehmer von Teil 2:
- Der Teilnehmer ist gegenüber einem Proteasom-Inhibitor refraktär, was definiert ist als Progression an oder innerhalb von 60 Tagen nach der letzten Dosis eines Behandlungsschemas mit einem Proteasominhibitor
- Behandlung mit einem Proteasom-Inhibitor innerhalb von 60 Tagen vor der ersten Dosis des Studienmedikaments
- Behandlung mit Antimyelom-Chemotherapie, Strahlentherapie, biologischer Therapie, Immuntherapie oder einer Forschungstherapie, einschließlich gezielter niedermolekularer Wirkstoffe innerhalb von 2 Wochen oder gegebenenfalls 5 Halbwertszeiten vor der ersten Dosis.
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt
