COM_STUDIES_DEFAULT_PAGE_TITLE
Dresden
Dresden
Dresden Universitätsklinikum Medizinische Klinik und Poliklinik I
Prof. Dr. Chr. Röllig, 0351 458 2321
Studien
AlloRelapseMM
Allogene Stammzelltransplantation im Vergleich zur konventionellen Therapie als Salvage-Therapie für Patienten mit rezidiviertem / progredientem Multiplen Myelom nach einer Erstlinientherapie (AlloRelapseMMStudy)
Organisatorische Daten:
| Prüfplancode: | |
| ISRCTN: | |
| EudraCT: | 2021-001--5-67 |
| Clinicaltrials.gov: | NCT0567319 |
| DRKS: | |
| Sponsor: | Universitätsklinikum Hamburg-Eppendorf mit Unterstützung des Gemeinsamen Bundesausschusses (G-BA) |
| Studienphase: | III |
| Status: | Rekrutierung läuft, geplant bis 6/2027 |
Ziel:
In dieser Studie wird untersucht, ob die Transplantation von gespendeten Stammzellen (allogene Stammzelltransplantation) Vorteile gegenüber der gängigen Therapie bei Patienten mit Multiplem Myelom bietet, bei denen es nach einer Transplantation von körpereigenen Stammzellen (autologe Stammzelltransplantation) als Erstlinientherapie zu einem Erkrankungsrückfall (Rezidiv) oder einem Fortschreiten der Krankheit (Progression) gekommen ist. Es wird geprüft, ob die allogene Stammzelltransplantation das Gesamtüberleben bei diesen Patienten verlängert.
Hintergrund:
Eine Behandlungsoption des Multiplen Myeloms ist die Stammzelltransplantation. Nach einer Chemotherapie, bei der die Krebszellen zerstört werden, werden körpereigene (autologe) oder gespendete körperfremde (allogene) Stammzellen übertragen. Die übertragenen Stammzellen siedeln sich dann im Knochenmark an und bilden „frische“ Blutzellen. Die Stammzelltransplantation kommt als Behandlungsoption nach der ersten Diagnose (Erstlinientherapie), aber auch bei einem Rückfall nach Vorbehandlung (jenseits der Erstlinientherapie) in Frage. Auch wenn das Multiple Myelom oft nicht endgültig heilbar ist, soll die Therapie die Lebenserwartung verlängern und die Symptome zurückdrängen. (Quelle: https://www.g-ba.de/studien/erprobung/allorelapsemmstudy-studie/)
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- Diagnose Multiples Myelom,
- Rückfall (Rezidiv) oder Fortschreiten (Progress) des Multiplen Myeloms unter oder nach einer autologen Stammzelltransplantation
- Erhalt von höchstens 1 Zyklus einer sogenannten Salvage-Therapie (Rezidiv-Therapie) vor Einschluss in die Studie
Ausschlusskriterien:
- Unzureichende Organfunktion gemäß der Definition im Prüfplan
- Aktive Hepatitis B oder C oder unkontrollierte HIV-Infektion
- Andere aktive bösartige Erkrankung
- Vorherige Behandlung mit allogenen Stammzellen
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Studie mit Iberdomid
Eine Studie zur Bestimmung der Dosis und Verträglichkeit der CC-220 Monotherapie mit Iberdomid, in Kombination mit Dexamethason und in Kombination mit Dexamethason und Daratumumab oder Bortezomib bei Patienten mit fortgeschrittenem und refraktärem Multiplem Myelom (RRMM)
Organisatorische Daten:
| Prüfplancode: | CC-220-MM-001 |
| ISRCTN: | |
| EudraCT: | 2016-000860-40 |
| Clinicaltrials.gov: | NCT02773030 |
| DRKS: | |
| Sponsor: | Celgene |
| Studienphase: | Phase 1b/2a |
| Status: | Rekrutierung läuft, geplant bis Mai 2026 |
Ziel:
Dies ist eine multizentrische, länderübergreifende, offene Phase-1b/2a-Dosiseskalationsstudie zur Bestimmung der höchsten verträglichen Dosis, der Sicherheit, Verträglichkeit, Pharmakokinetik (Einwirkung des Körpers auf die Substanz)und Wirksamkeit von CC-220 (Iberdomid) als Monotherapie und in Kombination mit anderen Therapien bei Patienten mit rezidivierendem oder therapierefraktärem Multiplem Myelom.
Hintergrund:
Iberdomid (CC-220) ist eine neuartige Substanz, die in dieser Phase I/II Studie zur Behandlung von fortgeschrittenem und refraktärem Multiplem Myelom in Kombination mit anderen Therapien untersucht wird. Präklinische Studien zu Iberdomid zeigen, dass es wirksamer an das Protein Cereblon bindet als andere Cereblon-bindende Substanzen. Klinische Studien zu Bortezomib und Daratumumab in Kombination mit anderen Cereblon-bindenden Mitteln haben eine hohe Verträglichkeit bei bemerkenswerter Wirksamkeit bei RRMM gezeigt. Allerdings wurden diese Kombinationen mit Iberdomid bisher nicht untersucht. Insgesamt gesehen untermauern die vorliegenden präklinischen Daten die Untersuchung von Iberdomid in Kombination mit sowohl Bortezomib/Dexamethason als auch Daratumumab in klinischen Studien.
(Michael Amatangelo et al, Blood 2018. 132, 1935; http://www.bloodjournal.org/content/132/Suppl_1/1935?sso-checked=true
Durchführung:
Die Studie wird in zwei Teilen durchgeführt: Teil 1 mit Kohorte A und B und Teil 2 mit Kohorte C, D, E, F, G1 und G2.
In Teil 1 (Phase 1b) wird die maximal verträgliche Dosis von oralem CC-220 als Monotherapie (Kohorte A) und die von oralem CC-220 in Kombination mit oralem Dexamethason (Kohorte B) bestimmt. Außerdem wird in diesem Teil die empfohlene CC-220-Dosis für Teil 2 (Phase 2) ermittelt.
In Teil 2 werden folgende Therapien untersucht:
Kohorte C: CC-220 als Monotherapie in der empfohlenen Dosis für Teil 2
Kohorte D: CC-220 in der empfohlenen Dosis für Teil 2 in Kombination mit oralem Dexamethason
Kohorte E:CC-220 in Kombination mit oralem Dexamethason und intravenösem Daratumumab
Kohorte F: CC-220 in Kombination mit oralem Dexamethason und subkutanem Bortezomib
Kohorte G1: CC-220 in Kombination mit einmal wöchentlich verabreichtem intravenösem Carfilzomib und oralem Dexamethason
Kohorte G2: CC-220 in Kombination mit zweimal wöchentlich verabreichtem intravenösem Carfilzomib und oralem Dexamethason.
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- Dokumentierte Diagnose eines Multiplen Myeloms (MM) und messbare Krankheit, definiert als
- M-Protein: Serum-Elektrophorese ≥ 0,5 g/dl und/oder Urinprotein-Elektrophorese ≥ 200 mg/24 Stunden und/oder
- Leichtketten-Myelom ohne messbare Erkrankung im Serum oder Urin - Dokumentierte Krankheitsprogression an oder innerhalb von 60 Tagen seit der letzten Dosis der letzten Myelombehandlung
- ECOG (Index zur Abstufung der Lebensqualität von Patienten mit Krebserkrankungen) von 0, 1 oder 2
Ausschlußkriterien:
- Nicht ekretorisches oder oligosekretorisches Multiples Myelom
- Plasmazellenleukämie oder Amyloidose
- Bestimmte Laborwerte, die nicht im Normalbereich liegen
- Periphere Neuropathie ≥ Grad 2
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Studie zur Untersuchung alternativer Dosierungsschemata von Belantamab-Mafodotin bei Teilnehmern mit rezidiviertem oder refraktärem
Multiplem Myelom (RRMM) (DREAMM 14)
Organisatorische Daten:
| Prüfplancode: | 209628 |
| ISRCTN: | |
| EudraCT: | 2021-004151-16 |
| Clinicaltrials.gov: | NCT05064358 |
| DRKS: | |
| Sponsor: | GlaxoSmithKline |
| Studienphase: | 2 |
| Status: | Rekrutierung geplant bis Mai 2024 |
Ziel:
Ziel dieser Studie ist es, alternative Dosierungsschemata von Belantamab-Mafodotin als Einzelwirkstoff bei Teilnehmern mit rezidiviertem oder refraktärem Multiplem Myelom (RRMM) zu untersuchen. So soll festgestellt werden, ob ein verbessertes Nutzen-/Risikoprofil insgesamt erreicht werden kann, wenn die Belantamab-Mafodotin-Dosis, der Zeitplan der Verabreichung oder beides geändert werden.
Hintergrund:
Belantamab-Mafodotin ist ein Antikörper-Wirkstoff-Konjugat, das zur Behandlung von Krebserkrankungen eingesetzt wird. Es ist richtet sich gegen das B-Zell-Reifungsantigen (BCMA).
BCMA ist auf der Oberfläche CD138-positiver Myelomzellen nachweisbar, nicht jedoch auf gesunden B-Zellen. Daher bietet sich BCMA als Zielstruktur in der Myelomtherapie an. Belantamab-Mafodotin ist für die Behandlung des Multiplen Myeloms zugelassen.
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- ECOG Performance Status (Index zur Abstufung der Lebensqualität von Patienten mit Krebserkrankungen) von 0 bis 2
- Der Patient hat eine histologisch oder zytologisch bestätigte Diagnose eines MM und
- a) hat sich zuvor einer Stammzelltransplantation unterzogen oder gilt als nicht transplantationsfähig, und
- b) hat auf mindestens drei vorherige Therapielinien für MM, einschließlich eines CD38-Antikörpers (z. B. Daratumumab) allein oder in Kombination, nicht angesprochen. Zudem ist der Patient refraktär gegenüber einem immunmodulatorischen Mittel (z. B. Lenalidomid, Pomalidomid) und einem Proteasom-Inhibitor (z. B. Bortezomib, Ixazomib, Carfilzomib).
- Der Teilnehmer hat eine messbare Erkrankung gemäß den modifizierten IMWG-Kriterien.
Ausschlußkriterien:
- Symptomatische Amyloidose, aktives POEMS-Syndrom (Polyneuropathie, Organomegalie, Endokrinopathie, Myelom-Protein und Hautveränderungen) oder aktive Plasmazellleukämie zum Zeitpunkt des Sreenings.
- Aktuelle Erkrankung des Hornhautepithels, ausgenommen nicht-konfluente, oberflächliche, punktförmige Keratitis (SPK).
- Anzeichen für aktive Schleimhaut- oder innere Blutungen.
- aktive Nierenerkrankung.
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Teclistamab, Daratumumab, Lenalidomid,Dexamethason +/- Bortezomib
Phase-2-Studie zur Bewertung der Sicherheit und Wirksamkeit von Teclistamab in Kombination mit Daratumumab, Lenalidomid und Dexamethason mit und ohne Bortezomib als Induktionstherapie und Teclistamab in Kombination mit Daratumumab und Lenalidomid als Erhaltungstherapie bei transplantationsgeeigneten Patienten mit neu diagnostiziertem Multiplem Myelom
Organisatorische Daten:
| Prüfplancode: | GMMG-HD10, DSMM-XX, 64007957MMY2003, MajesTEC-5 |
| ISRCTN: | |
| EudraCT: | |
| Clinicaltrials.gov: | NCT05695508 |
| DRKS: | |
| Sponsor: | Universitätsklinikum Heidelberg in Zusammenarbeit mit Janssen |
| Studienphase: | Phase 3 |
| Status: | Rekrutierung läuft, geplant bis Mai 2026 |
Hintergrund:
Teclistamab ist ein bispezifischer Antikörper, der an das B-Zell-Reifungsantigen (BCMA), ein Protein auf der Oberfläche von Myelomzellen, bindet und gegen CD3-Rezeptoren auf der Oberfläche von T-Zellen gerichtet ist. Teclistamab wurde im August 2022 in der EU als Monotherapie zur Behandlung des fortgeschrittenen Multiplen Myeloms zugelassen und wird jetzt bei weiteren Indikationen geprüft.
In dieser Studie bei Patienten mit neu diagnostiziertem Multiplem Myelom, die für eine Transplantation geeignet sind, werden die Sicherheit, Verträglichkeit und Wirksamkeit einer Fünffach-Kombination in der Induktionstherapie geprüft: Teclistamab (Tec) in Kombination mit Daratumumab (Dara), Lenalidomid (R), Bortezomib (V) und Dexamethason (d).
Durchführung
Das Studiendesign gliedert sich in drei Studienarme:
Patienten in Arm A und B erhalten eine Induktionstherapie mit Tec-Dara-Rd (Arm A) oder Tec-Dara-VRd (Arm B). Es folgen eine Hochdosis-Chemotherapie und eine autologe Stammzelltransplantation gemäß Therapiestandard sowie eine Erhaltungstherapie mit Tec-Dara-R.
Patienten in Arm C erhalten eine Induktionstherapie gemäß Therapiestandard gefolgt von einer Hochdosis-Chemotherapie und einer autologen Stammzelltransplantation sowie gegebenenfalls einer Konsolidierungstherapie. Im Anschluss daran folgt eine Erhaltungstherapie mit Tec-Dara-R.
Die Behandlungsdauer der Induktionsphase beträgt 6 Zyklen (à 28 Tage). Im Anschluss daran folgen eine Hochdosis-Chemotherapie und autologe Stammzelltransplantation . Danach beginnt die Erhaltungsphase der Studie unter Tec-Dara-R über 18 Zyklen.
Studienarm C beginnt mit dem Screening nach abgeschlossener Hochdosis-Chemotherapieplus autologer Stammzelltransplantation. Die Behandlung erfolgt genauso wie in Arm A und B mit Tec-Dara-R als Erhaltungstherapie für 18 Zyklen.
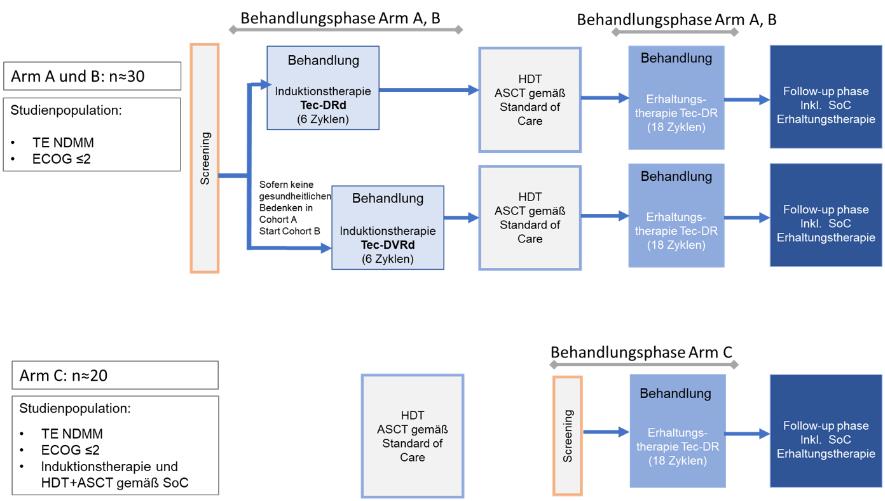
Abb. Studiendesign, mit freundlicher Genehmigung des Universitätsklinikums Heidelberg, Prof. Raab
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- Neu diagnostiziertes Multiples Myelom gemäß den Diagnosekriterien der International Myeloma Working Group (IMWG) und Erhalt einer Induktionstherapie mit oder ohne Konsolidierung
- Ein ECOG-Performance-Status-Score (Index zur Abstufung der Lebensqualität von Patienten mit Krebserkrankungen) von 0, 1 oder 2 beim Screening und unmittelbar vor Beginn der Verabreichung der Studienbehandlung
- Messbare Erkrankung während der Screening-Phase, gemäß der Definition im Protokoll
Ausschlußkriterien
- Beteiligung des ZNS oder klinische Anzeichen für eine Beteiligung der Hirnhäute
- Schlaganfall oder Krampfanfall innerhalb von 6 Monaten vor Studienbeginn
- Transplantation in der Krankengeschichte, die eine immunsuppressive Behandlung erforderte
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Induktionstherapie mit Lenalidomid, Bortezomib und Dexamethason und entweder intravenösem oder subkutanem Isatuximab bei Patienten mit neu diagnostiziertem Multiplem Myelom, die für eine Hochdosis-Chemotherapie mit nachfolgender autologer Stammzelltransplantation geeignet sind
Organisatorische Daten:
| Prüfplancode: | GMMG-HD 8 / DSMM XIX |
| ISRCTN: | |
| EudraCT: | 2022-000996-38 |
| Clinicaltrials.gov: | NCT05804032 |
| DRKS: | |
| Sponsor: | Universitätsklinikum Heidelberg DSMM, Sanofi und KKS-Netzwerk |
| Studienphase: | |
| Status: | Rekrutierung geplant bis Juni 2025 |
Ziel:
In dieser Studie für Patienten mit zuvor unbehandeltem Multiplem Myelom soll die Wirksamkeit der Induktionstherapie mit Isatuximab in Kombination mit Lenalidomid/Bortezomib/Dexamethason (RVd) bei Gabe von Isatuximab unter die Haut (subkutan) gegenüber einer Gabe in die Vene (intravenös) untersucht werden.
Durchführung:
Randomisierung: Die Patienten werden vor der Induktionstherapie nach dem Zufallsverfahren (randomisiert) einem von 2 Studienarmen (A oder B) zugeordnet.
- Patienten in Arm A erhalten 3 Zyklen des monoklonalen Antikörpers Isatuximab intravenös in Kombination mit dem RVd-Schema (Lenalidomid, Bortezomib, Dexamethason). Jeder Zyklus hat eine Dauer von 42 Tagen.
- Patienten in Arm B erhalten 3 Zyklen Isatuximab subkutan in Kombination mit RVd.
Nach der Induktionstherapie erhalten die Patienten eine Standard-Intensivierung (in der Regel eine Mobilisierungstherapie auf Cyclophosphamid-Basis, Stammzellenentnahme und hochdosiertes Melphalan, gefolgt von einer autologen Stammzelltransplantation
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- Bestätigte Diagnose eines neu diagnostizierten behandlungsbedürftigen Multiplen Myeloms gemäß den überarbeiteten Diagnosekriterien der International Myeloma Working Group (IMWG))
- Der Patient ist für eine Hochdosis-Chemotherapie und eine autologe Stammzelltransplantation geeignet
- Messbare Krankheitsaktivität (gemäß der Definition im Prüfplan)
- Alter 18 bis 70 Jahre bei Studieneinschluss
Ausschlußkriterien:
- Bekannte Überempfindlichkeit (oder Kontraindikation) gegen einen der Bestandteile der Studientherapie
- Systemische Amyloid-Leichtketten-Amyloidose (außer lokalisierte Amyloid-Leichtketten -Amyloidose, die auf die Haut oder das Knochenmark beschränkt ist)
- Plasmazell-Leukämie
- Vorherige Chemo- oder Strahlentherapie in den letzten 5 Jahren (außer lokaler Strahlentherapie bei lokalem Fortschreiten des Myeloms)
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Dies ist eine randomisierte, multizentrische, offene Phase-3-Studie zur Bewertung der subkutanen (SC) gegenüber der intravenösen (IV) Verabreichung von Isatuximab in Kombination mit Pomalidomid und Dexamethason (Pd) bei Patienten mit rezidivierendem refraktärem Multiplen Myelom (RRMM), die mindestens eine vorherige Therapielinie mit Lenalidomid und einem Proteasominhibitor (PI) erhalten haben.
Organisatorische Daten:
| Prüfplancode: | EFC15951 |
| ISRCTN: | |
| EudraCT: | 2021-002485-41 |
| Clinicaltrials.gov: | NCT05405166 |
| DRKS: | |
| Sponsor: | Sanofi |
| Studienphase: | Phase 3 |
| Status: | Rekrutierung läuft, geplant bis Mai 2024 |
Durchführung:
Die in Frage kommenden Teilnehmer werden im Verhältnis 1:1 nach dem Zufallsprinzip (randomisiert) einem von 2 Studienarmen zugeordnet:
Arm SC: Isatuximab SC + Pd
Arm IV: Isatuximab IV + Pd
Die Teilnehmer dürfen die Therapie so lange fortsetzen, bis die Krankheit fortschreitet, inakzeptable unerwünschte Ereignisse auftreten, der Teilnehmer den Abbruch der Therapie wünscht oder ein anderer Grund vorliegt, je nachdem, was zuerst eintritt.
Hintergrund:
Isatuximab ist ein gegen Tumoren gerichteter Wirkstoff und besitzt eine abtötende (zytotoxische) Wirkung auf die Myelom-Zellen. Es gehört zur Gruppe der monoklonalen Antikörper für die Behandlung des Multiplen Myeloms bei Erwachsenen. Die Wirkung von Isatuximab beruht auf der Bindung an den CD38-Rezeptor, was zum Zelltod führt.
Isatuximab wird ist als Therapie des rezidivierenden und refraktären Multiplen Myeloms bei Patienten angewendet, die bereits wegen ihres Multiplen Myeloms behandelt wurden. Es wird gemeinsam mit zwei verschiedenen Arzneimittelkombinationen angewendet:
- Pomalidomid und Dexamethason oder
- Carfilzomib und Dexamethason.
Bisher wird Isatuximab als Tropfinfusion in eine Vene verabreicht (intravenöse Infusion). In dieser Studie wird Isatuximab als Injektion unter die Haut (subkutan) geprüft
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien
- Teilnehmer mit Multiplem Myelom, die mindestens eine vorherige Therapielinie mit Lenalidomid und einem Proteasom-Inhibitor erhalten haben
- Teilnehmer mit messbarer Erkrankung definiert als
- M-Protein im Serum (≥ 0,5 g/dl) und/oder
- M-Protein im Urin (≥ 200 mg/24 Stunden) und/oder
- freie Leichtketten (FLC) im Serum (≥10 mg/dl und anormales Serum-FLC-Verhältnis
Ausschlußkriterien:
- Teilnehmer unter 18 Jahren
- Teilnehmer mit primär refraktärem Multiplem Myelom
- Teilnehmer, die nicht auf Anti-CD38 ansprechen bei einer Auswaschphase von weniger als 9 Monaten, oder die monoklonale Anti-CD38-Antikörper nicht vertragen
- Teilnehmer, die vorher mit Pomalidomid behandelt wurden
- Teilnehmer mit einem ECOG (Index zur Abstufung der Lebensqualität von Patienten mit Krebserkrankungen) > 2
- Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Eine Studie zum Vergleich von Teclistamab-Monotherapie mit Pomalidomid, Bortezomib, Dexamethason (PVd) oder Carfilzomib, Dexamethason (Kd) bei Teilnehmern mit rezidiviertem oder refraktärem Multiplem Myelom (MajesTEC-9)
Organisatorische Daten:
| Prüfplancode: | CR109244, 64007957MMY3006 |
| ISRCTN: | |
| EudraCT: | 2022-000928-37 |
| Clinicaltrials.gov: | NCT05572515 |
| DRKS: | |
| Sponsor: | Janssen |
| Studienphase: | Phase 3 |
| Status: | Rekrutierung läuft, geplant bis August 2025 |
Ziele:
Ziel dieser Studie ist es, die Wirksamkeit von Teclistamab mit PVd bzw. Kd bei Patienten mit rezidiviertem oder refraktärem Multiplem Myelom zu vergleichen, die bereits 1 bis 3 Therapielinien einschließlich einen monoklonalen Anti-CD38-Antikörpers und Lenalidomid erhalten haben.
Hintergrund:
Teclistamab ist ein bispezifischer Antikörper, der sowohl gegen das B-Zell-Reifungsantigen (BCMA), ein Protein auf Myelomzellen, als auch gegen CD3-Rezeptoren auf der T-Zell-Oberfläche gerichtet ist. Teclistamab wurde im August 2022 in der EU als Monotherapie zur Behandlung des fortgeschrittenen Multiplen Myeloms zugelassen und wird jetzt bei weiteren Indikationen geprüft.
In der MajesTEC-1-Studie bei stark vorbehandelten Patienten mit Multiplem Myelom (n=165) bewirkte Teclistamab ein hohes und dauerhaftes Ansprechen. Bei einer medianen Nachbeobachtungszeit von etwa 14 Monaten betrug die Gesamtansprechrate 63 Prozent, wobei 39,4 Prozent ein komplettes Ansprechen oder besser erreichten. Diese Daten wurden auf der Jahrestagung 2022 der American Society of Clinical Oncology (ASCO) veröffentlicht.
Durchführung:
Die Studie umfasst eine Screeningphase, eine Behandlungsphase und eine Nachbeobachtungsphase. Die Sicherheit wird durch körperliche Untersuchungen, neurologische Untersuchungen, den ECOG-Leistungsstatus (Eastern Cooperative Oncology Group), klinische Labortests, Vitalparameter und die Überwachung von Nebenwirkungen beurteilt. Die Gesamtdauer der Studie wird bis zu 9 Jahre betragen.
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- Dokumentierte Diagnose eines multiplen Myeloms gemäß den nachstehenden Kriterien: (a) Diagnose des Multiplen Myeloms gemäß den Diagnosekriterien der International Myeloma Working Group (IMWG) (b) Messbare Erkrankung zum Zeitpunkt des Screenings, gemäß der Definition im Protokoll. 1 bis 3 vorangegangene Therapielinien gegen das Myelom, einschließlich mindestens 2 aufeinanderfolgender Zyklen eines monoklonalen Antikörpers gegen Cluster of Differentiation 38 (CD38) in der zugelassenen Dosierung in einer beliebigen vorangegangenen Therapielinie und 2 aufeinanderfolgende Zyklen von Lenalidomid in einer beliebigen vorangegangenen Therapielinie.
- Dokumentierte Anzeichen für ein Fortschreiten der Krankheit oder das Ausbleiben eines Ansprechens auf die letzte Therapielinie, basierend auf der Bestimmung des Ansprechens durch den Prüfarzt nach den Kriterien der IMWG.
- ECOG Performance Status (Index zur Abstufung der Lebensqualität von Patienten mit Krebserkrankungen) von 0 bis 2.
Ausschlusskriterien:
- Eine vorherige gegen das B-Zell-Reifungsantigen (BCMA)-gerichtete Therapie
- Teilnehmer, für den PVd als Kontrolltherapie aus im Protokoll definierten Gründen nicht infrage kommt.
- Teilnehmer, für den Kd als Kontrolltherapie aus im Protokoll definierten Gründen nicht infrage kommt.
- Beteiligung des zentralen Nervensystems (ZNS) oder klinische Anzeichen einer meningealen Beteiligung des Multiplen Myeloms.
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt
Eine Studie mit Talquetamab und Teclistamab jeweils in Kombination mit einem PD-1-Inhibitor (Programmed Cell Death Receptor-1) zur Behandlung von Teilnehmern mit rezidiviertem oder refraktärem Multiplem Myelom (TRIMM-3)
Organisatorische Daten:
| Prüfplancode: | CR109168, 64407564MMY1005 |
| ISRCTN: | |
| EudraCT: | 2021-005073-22 |
| Clinicaltrials.gov: | NCT05338775 |
| DRKS: | |
| Sponsor: | Janssen |
| Studienphase: | 1 |
| Status: | Rekrutierung läuft, geplant bis Sept. 2024 |
Ziel:
Ziel der Studie ist es, die sichere Dosis bzw. sicheren Dosen eines PD-1-Inhibitors in Kombination mit Talquetamab oder Teclistamab zu ermitteln und die Sicherheit und Verträglichkeit von Talquetamab oder Teclistamab bei Verabreichung in Kombination mit einem PD-1-Inhibitor zu charakterisieren.
Hintergrund:
Talquetamab ist ein humanisierter monoklonaler Antikörper, der gegen zwei Ziele gerichtet (bispezifisch) ist: gegen humanes CD3, ein Oberflächenantigen auf T-Zellen, und gegen GPRC5D, ein Antigen in Tumorzellen oder auf deren Oberfläche (tumor-assoziiertes Antigen). Bei Verabreichung bindet Talquetamab sowohl an CD3 auf T-Zellen als auch an GPRC5D, das auf bestimmten Tumorzellen wie z.B. dem Multiplen Myelom vermehrt vorkommt und eine Schlüsselrolle bei der Vermehrung von Tumorzellen spielt.
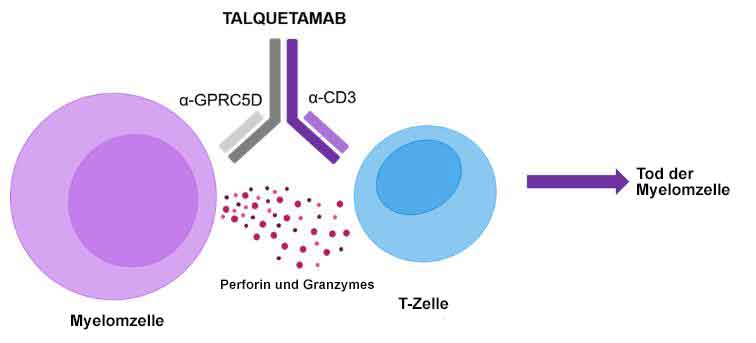
Abb. aus https://multiplemyelomahub.com/,
Teclistamab (JNJ-7957) ist ein bispezifischer Antikörper, der sowohl gegen BCMA als auch gegen CD3 gerichtet ist. BCMA ist ein Zelloberflächenprotein, das besonders auf Myelom- und Plasmazellen vorkommt und eine besondere biologische Bedeutung für das Überleben der Plasmazelle hat. Es wird bei Menschen mit Multiplem Myelom in signifikant höherem Maße exprimiert. CD3 ist an der Aktivierung der Immunantwort zur Bekämpfung von Infektionen beteiligt, Teclistamab lenkt CD3-T-Zellen auf BCMA-exprimierende Myelomzellen um, um die Zytotoxizität der Zielzellen zu induzieren. Ergebnisse aus präklinischen Studien zeigen, dass Teclistamab Myelom-Zelllinien und Myelom-Knochenmarkzellen von stark vorbehandelten Patienten abtötet.
Diese Studie geht davon aus, dass Talquetamab oder Teclistamab in Kombination mit einem PD-1-Inhibitor bei der Behandlung von rezidiviertem oder refraktärem Multiplem Myelom zu einem verbesserten klinischen Ansprechen führen, was auf verschiedene Wirkmechanismen zurückzuführen ist.
Einschlusskriterien:
- Dokumentierte Erstdiagnose eines Multiplen Myeloms nach den Diagnosekriterien der Internationalen Myelom-Arbeitsgruppe (IMWG)
- Teilnehmer mit rezidivierender oder refraktärer Erkrankung, die nicht für eine verfügbare Therapie mit nachgewiesenem klinischen Nutzen infrage kommen.
- Messbare Erkrankung zum Zeitpunkt des Screenings
- ECOG (Index zur Abstufung der Lebensqualität von Patienten mit Krebserkrankungen) von 0 oder 1
Ausschlusskriterien:
-
Vorangegangene Antitumortherapie innerhalb von 21 Tagen vor der ersten Dosis der Studienbehandlung (Therapie mit Proteasom-Inhibitoren [PI] oder Strahlentherapie innerhalb von 14 Tagen, Therapie mit immunmodulatorischen Wirkstoffen (IMiD) innerhalb von 7 Tagen, genmodifizierte adoptive Zelltherapie oder autologe Stammzelltransplantation innerhalb von 3 Monaten)
-
Vorherige Therapie mit PD-1-Inhibitoren, allogene Stammzelltransplantation oder Organtransplantation
-
Aktive Plasmazellleukämie, Waldenstrom-Makroglobulinämie, POEMS-Syndrom (Polyneuropathie, Organomegalie, Endokrinopathie, M-Protein und Hautveränderungen) oder primäre Leichtkettenamyloidose
-
Aktive Beteiligung des zentralen Nervensystems (ZNS) oder klinische Anzeichen einer meningealen Beteiligung des Multiplen Myeloms. Bei Verdacht auf eine der beiden Erkrankungen sind eine Magnetresonanztomographie des Gehirns (MRT) und eine Lumbalzytologie erforderlich.
-
Impfung mit einem abgeschwächten Lebendimpfstoff innerhalb von 4 Wochen vor der ersten Dosis der Studienbehandlung
Alle weiteren Kritereien besprechen Sie mit Ihrem Arzt
Gemeinschaftspraxis Hämatologie - Onkologie
Dr. Illmer, 0351-44 00022
Studien
DREAMM-9Belantamab-Mafodotin in Kombination mit Bortezomib + Lenalidomid + Dexamethason
Organisatorische Daten:
| Prüfplancode: | 209664 |
| ISRCTN: | |
| EudraCT: | 2019-003047-30 |
| Clinicaltrials.gov: | NCT04091126 |
| DRKS: | |
| Sponsor: | GlaxoSmithKline |
| Studienphase: | I |
| Status: | Rekrutierung geplant bis 11/2025 |
Ziel:
In dieser randomisierten, offenen Phase-I-Studie werden die Wirksamkeit und Sicherheit von Belantamab-Mafodotin in Kombination mit Bortezomib, Lenalidomid und Dexamethason (VRd) im Vergleich zu VRd allein bei Patienten mit neu neudiagnostiziertem Multiplen Myelom untersucht, die nicht für eine autologe Stammzelltransplantation geeignet sind.
Durchführung:
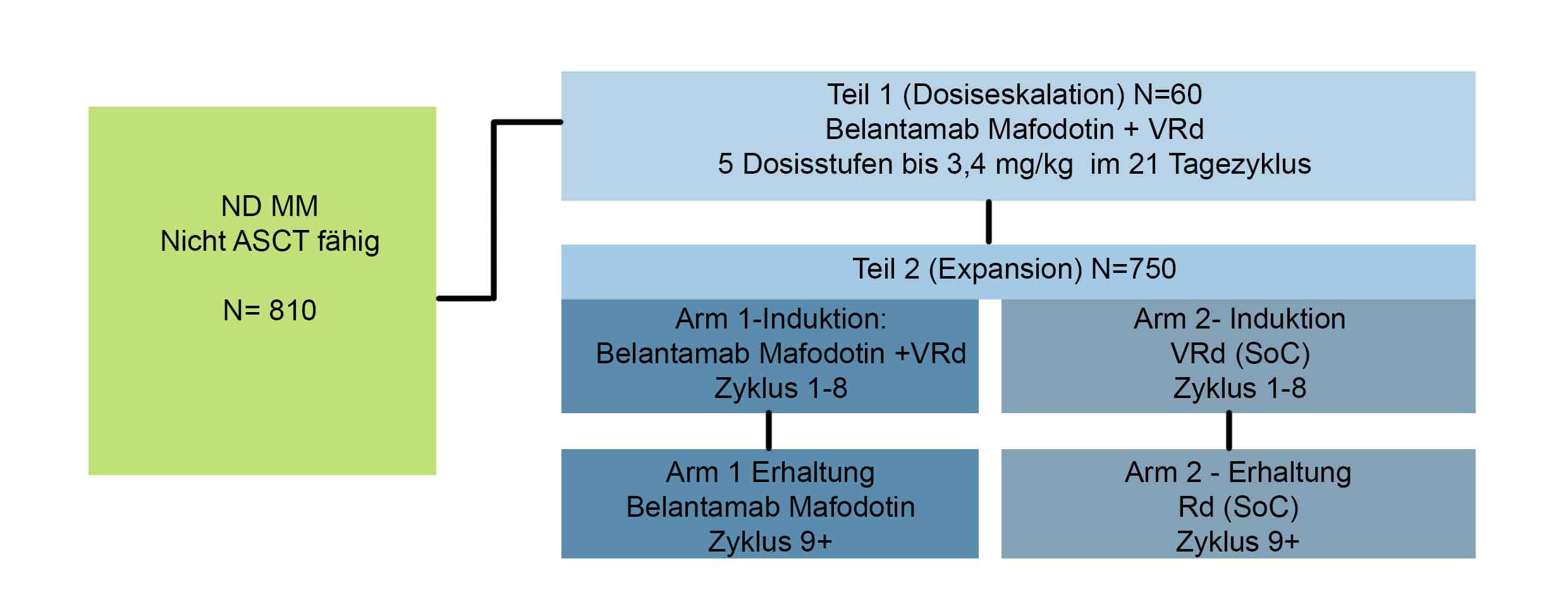
Die Studie besteht aus zwei Teilen:
In Teil 1 werden die Sicherheit und Verträglichkeit von Belantamab-Mafodotin in bis zu 5 Dosisstufen in Kombination mit VRd bewertet und die empfohlene Dosis für die Phase III (Teil 2) ermittelt.
In Teil 2 (Verlängerung) werden die Wirksamkeit und Sicherheit der für die Phase III empfohlenen Dosis von Belantamab-Mafodotin in Kombination mit VRd im Vergleich zu VRd allein bewertet.
Hintergrund:
Belantamab-Mafodotin (vormals GSK2857916) ist ein gegen das Oberflächenantigen BCMA (engl. B-Cell Maturation Antigen) gerichtetes Antikörper-Wirkstoff-Konjugat. Sobald Belantamab an BCMA bindet, wird das krebstötende Molekül in die Zelle freigesetzt und führt den Zelltod herbei. Bei stark vortherapierten Patienten mit rezidiviertem/refraktärem multiplem Myelom hat Belantamab-Mafodotin eine hohe Wirksamkeit gezeigt. Weitere Untersuchungen müssen zeigen, inwieweit auftretende Nebenwirkungen, teilweise mit vorübergehender Beeinträchtigung des Sehvermögens, die Einsatzmöglichkeiten dieser Therapie einschränken. Vielversprechend erscheinen Kombinationen mit etablierten Substanzen zur weiteren Verbesserung der Ansprechrate, -tiefe, und -dauer.
Belantamab-Mafodotin ist noch nicht zur Anwendung zugelassen. Allerdings wird der Antrag auf Zulassung des Arzneimittels für die Behandlung von Patienten mit rezidiviertem oder therapierefraktärem Multiplen Myelom im Rahmen eines beschleunigten Verfahrens derzeit sowohl von der amerikanischen Zulassungsbehörde FDA als auch der europäischen Arzneimittelbehörde EMA geprüft. (Stand der Information: Februar 2020, https://myelomaresearchnews.com/2020/02/05/ema-review-gsks-belantamab-mafodotin-treatment-heavily-pretreated-multiple-myeloma/
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
-
ECOG Performance Status (Index zur Abstufung der Lebensqualität von Patienten mit Krebserkrankungen) von 0 bis2
-
Neudiagnose Multiples Myelom
-
Der Patient ist für die Hochdosistherapie und die autologe Stammzelltransplantation nicht geeignet
-
Der Patient erfüllt mindestens eines der folgenden Kriterien einer messbaren Krankheitsaktivität:
Urin-Leichtkette (M-Protein) ≥ 200 mg/24 Stunden ≥ 0,2 g/24 Stunden) oder
Serum M-Protein ≥ 0,5 g/l ≥ 5,0 g/l]) oder
Konzentration der betroffenen freien Leichtkette im Serum (sFLC) ≥ 10 mg/dl (≥ 100 mg/l) und abnormes sFLC-Verhältnis (< 0,26 oder > 1,65).
Ausschlußkriterien:
- Smouldering Myelom (SMM)
- Systemische Vortherapie (mit Ausnahme von insgesamt bis zu 160 mg Dexamethason über 4 Tage)
- Periphere Neuropathie ≥ Grad
- Größere Operation innerhalb von 4 Wochen vor der ersten Gabe der Studienmedikation
- Keratopathie
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Gemeinschaftspraxis Mohm/Prange-Krex
0351-4416018
Onkozentrum Dresden / Freiberg
0351- 795 255 0
