COM_STUDIES_DEFAULT_PAGE_TITLE
Rheinland-Pfalz
Kaiserslautern
Westpfalz-Klinikum GmbH
0631-203-0
Koblenz
Gemeinschaftsklinikum Ev. Stift. St. Martin
Studien
GMMG-HD8/DSMM-XIX
Induktionstherapie mit Lenalidomid, Bortezomib und Dexamethason und entweder intravenösem oder subkutanem Isatuximab bei Patienten mit neu diagnostiziertem Multiplem Myelom, die für eine Hochdosis-Chemotherapie mit nachfolgender autologer Stammzelltransplantation geeignet sind
Organisatorische Daten:
| Prüfplancode: | GMMG-HD 8 / DSMM XIX |
| ISRCTN: | |
| EudraCT: | 2022-000996-38 |
| Clinicaltrials.gov: | NCT05804032 |
| DRKS: | |
| Sponsor: | Universitätsklinikum Heidelberg DSMM, Sanofi und KKS-Netzwerk |
| Studienphase: | |
| Status: | Rekrutierung geplant bis Juni 2025 |
Ziel:
In dieser Studie für Patienten mit zuvor unbehandeltem Multiplem Myelom soll die Wirksamkeit der Induktionstherapie mit Isatuximab in Kombination mit Lenalidomid/Bortezomib/Dexamethason (RVd) bei Gabe von Isatuximab unter die Haut (subkutan) gegenüber einer Gabe in die Vene (intravenös) untersucht werden.
Durchführung:
Randomisierung: Die Patienten werden vor der Induktionstherapie nach dem Zufallsverfahren (randomisiert) einem von 2 Studienarmen (A oder B) zugeordnet.
- Patienten in Arm A erhalten 3 Zyklen des monoklonalen Antikörpers Isatuximab intravenös in Kombination mit dem RVd-Schema (Lenalidomid, Bortezomib, Dexamethason). Jeder Zyklus hat eine Dauer von 42 Tagen.
- Patienten in Arm B erhalten 3 Zyklen Isatuximab subkutan in Kombination mit RVd.
Nach der Induktionstherapie erhalten die Patienten eine Standard-Intensivierung (in der Regel eine Mobilisierungstherapie auf Cyclophosphamid-Basis, Stammzellenentnahme und hochdosiertes Melphalan, gefolgt von einer autologen Stammzelltransplantation
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- Bestätigte Diagnose eines neu diagnostizierten behandlungsbedürftigen Multiplen Myeloms gemäß den überarbeiteten Diagnosekriterien der International Myeloma Working Group (IMWG))
- Der Patient ist für eine Hochdosis-Chemotherapie und eine autologe Stammzelltransplantation geeignet
- Messbare Krankheitsaktivität (gemäß der Definition im Prüfplan)
- Alter 18 bis 70 Jahre bei Studieneinschluss
Ausschlußkriterien:
- Bekannte Überempfindlichkeit (oder Kontraindikation) gegen einen der Bestandteile der Studientherapie
- Systemische Amyloid-Leichtketten-Amyloidose (außer lokalisierte Amyloid-Leichtketten -Amyloidose, die auf die Haut oder das Knochenmark beschränkt ist)
- Plasmazell-Leukämie
- Vorherige Chemo- oder Strahlentherapie in den letzten 5 Jahren (außer lokaler Strahlentherapie bei lokalem Fortschreiten des Myeloms)
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
InVo- Institut für Versorgungsforschung in der Onkologie
0261-92156930
Praxisklinik für Haematologie und Onkologie Koblenz
0261 30493-0, 0261 30493-33
Studien
DREAMM-9Belantamab-Mafodotin in Kombination mit Bortezomib + Lenalidomid + Dexamethason
Organisatorische Daten:
| Prüfplancode: | 209664 |
| ISRCTN: | |
| EudraCT: | 2019-003047-30 |
| Clinicaltrials.gov: | NCT04091126 |
| DRKS: | |
| Sponsor: | GlaxoSmithKline |
| Studienphase: | I |
| Status: | Rekrutierung geplant bis 11/2025 |
Ziel:
In dieser randomisierten, offenen Phase-I-Studie werden die Wirksamkeit und Sicherheit von Belantamab-Mafodotin in Kombination mit Bortezomib, Lenalidomid und Dexamethason (VRd) im Vergleich zu VRd allein bei Patienten mit neu neudiagnostiziertem Multiplen Myelom untersucht, die nicht für eine autologe Stammzelltransplantation geeignet sind.
Durchführung:
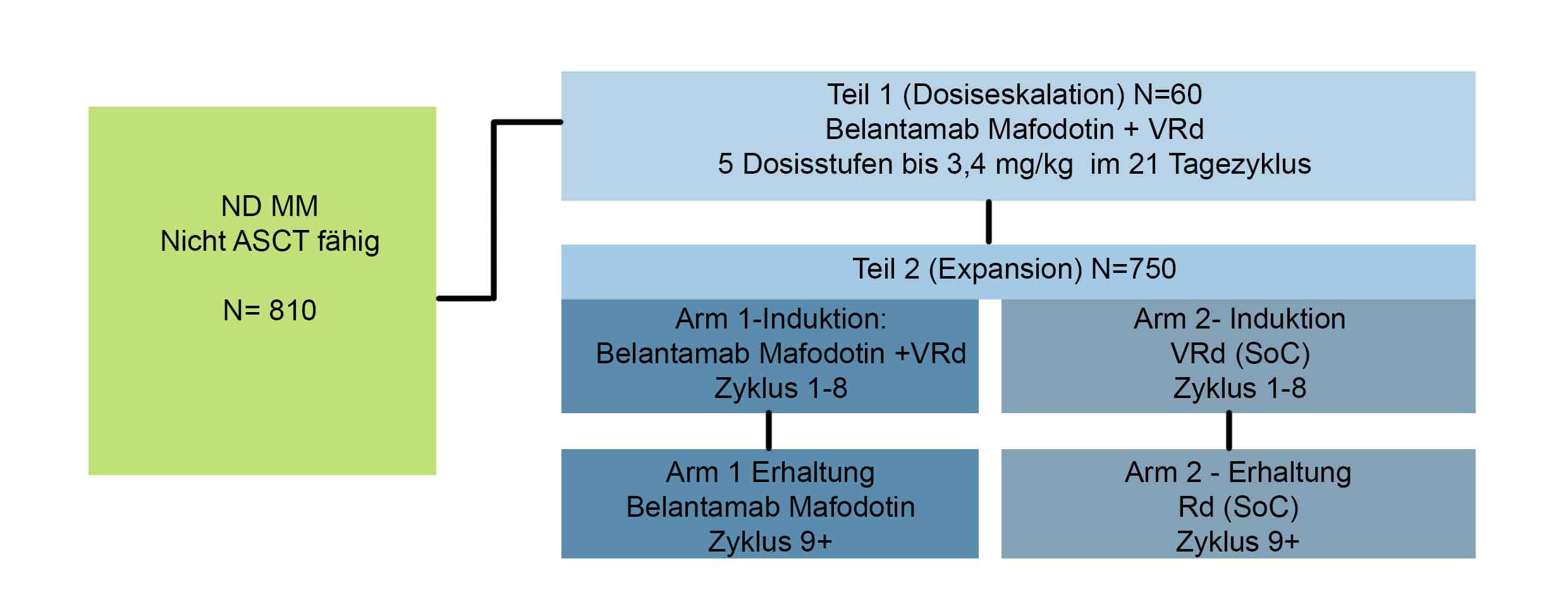
Die Studie besteht aus zwei Teilen:
In Teil 1 werden die Sicherheit und Verträglichkeit von Belantamab-Mafodotin in bis zu 5 Dosisstufen in Kombination mit VRd bewertet und die empfohlene Dosis für die Phase III (Teil 2) ermittelt.
In Teil 2 (Verlängerung) werden die Wirksamkeit und Sicherheit der für die Phase III empfohlenen Dosis von Belantamab-Mafodotin in Kombination mit VRd im Vergleich zu VRd allein bewertet.
Hintergrund:
Belantamab-Mafodotin (vormals GSK2857916) ist ein gegen das Oberflächenantigen BCMA (engl. B-Cell Maturation Antigen) gerichtetes Antikörper-Wirkstoff-Konjugat. Sobald Belantamab an BCMA bindet, wird das krebstötende Molekül in die Zelle freigesetzt und führt den Zelltod herbei. Bei stark vortherapierten Patienten mit rezidiviertem/refraktärem multiplem Myelom hat Belantamab-Mafodotin eine hohe Wirksamkeit gezeigt. Weitere Untersuchungen müssen zeigen, inwieweit auftretende Nebenwirkungen, teilweise mit vorübergehender Beeinträchtigung des Sehvermögens, die Einsatzmöglichkeiten dieser Therapie einschränken. Vielversprechend erscheinen Kombinationen mit etablierten Substanzen zur weiteren Verbesserung der Ansprechrate, -tiefe, und -dauer.
Belantamab-Mafodotin ist noch nicht zur Anwendung zugelassen. Allerdings wird der Antrag auf Zulassung des Arzneimittels für die Behandlung von Patienten mit rezidiviertem oder therapierefraktärem Multiplen Myelom im Rahmen eines beschleunigten Verfahrens derzeit sowohl von der amerikanischen Zulassungsbehörde FDA als auch der europäischen Arzneimittelbehörde EMA geprüft. (Stand der Information: Februar 2020, https://myelomaresearchnews.com/2020/02/05/ema-review-gsks-belantamab-mafodotin-treatment-heavily-pretreated-multiple-myeloma/
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
-
ECOG Performance Status (Index zur Abstufung der Lebensqualität von Patienten mit Krebserkrankungen) von 0 bis2
-
Neudiagnose Multiples Myelom
-
Der Patient ist für die Hochdosistherapie und die autologe Stammzelltransplantation nicht geeignet
-
Der Patient erfüllt mindestens eines der folgenden Kriterien einer messbaren Krankheitsaktivität:
Urin-Leichtkette (M-Protein) ≥ 200 mg/24 Stunden ≥ 0,2 g/24 Stunden) oder
Serum M-Protein ≥ 0,5 g/l ≥ 5,0 g/l]) oder
Konzentration der betroffenen freien Leichtkette im Serum (sFLC) ≥ 10 mg/dl (≥ 100 mg/l) und abnormes sFLC-Verhältnis (< 0,26 oder > 1,65).
Ausschlußkriterien:
- Smouldering Myelom (SMM)
- Systemische Vortherapie (mit Ausnahme von insgesamt bis zu 160 mg Dexamethason über 4 Tage)
- Periphere Neuropathie ≥ Grad
- Größere Operation innerhalb von 4 Wochen vor der ersten Gabe der Studienmedikation
- Keratopathie
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Ludwigshafen
Klinikum der Stadt Ludwigshafen am Rhein GmbH
0621-503-3994
Mainz
III. Medizinische Klinik und Poliklinik, Johannes Gutenberg-Universität
06131-17-0
Studien
AlloRelapseMM
Allogene Stammzelltransplantation im Vergleich zur konventionellen Therapie als Salvage-Therapie für Patienten mit rezidiviertem / progredientem Multiplen Myelom nach einer Erstlinientherapie (AlloRelapseMMStudy)
Organisatorische Daten:
| Prüfplancode: | |
| ISRCTN: | |
| EudraCT: | 2021-001--5-67 |
| Clinicaltrials.gov: | NCT0567319 |
| DRKS: | |
| Sponsor: | Universitätsklinikum Hamburg-Eppendorf mit Unterstützung des Gemeinsamen Bundesausschusses (G-BA) |
| Studienphase: | III |
| Status: | Rekrutierung läuft, geplant bis 6/2027 |
Ziel:
In dieser Studie wird untersucht, ob die Transplantation von gespendeten Stammzellen (allogene Stammzelltransplantation) Vorteile gegenüber der gängigen Therapie bei Patienten mit Multiplem Myelom bietet, bei denen es nach einer Transplantation von körpereigenen Stammzellen (autologe Stammzelltransplantation) als Erstlinientherapie zu einem Erkrankungsrückfall (Rezidiv) oder einem Fortschreiten der Krankheit (Progression) gekommen ist. Es wird geprüft, ob die allogene Stammzelltransplantation das Gesamtüberleben bei diesen Patienten verlängert.
Hintergrund:
Eine Behandlungsoption des Multiplen Myeloms ist die Stammzelltransplantation. Nach einer Chemotherapie, bei der die Krebszellen zerstört werden, werden körpereigene (autologe) oder gespendete körperfremde (allogene) Stammzellen übertragen. Die übertragenen Stammzellen siedeln sich dann im Knochenmark an und bilden „frische“ Blutzellen. Die Stammzelltransplantation kommt als Behandlungsoption nach der ersten Diagnose (Erstlinientherapie), aber auch bei einem Rückfall nach Vorbehandlung (jenseits der Erstlinientherapie) in Frage. Auch wenn das Multiple Myelom oft nicht endgültig heilbar ist, soll die Therapie die Lebenserwartung verlängern und die Symptome zurückdrängen. (Quelle: https://www.g-ba.de/studien/erprobung/allorelapsemmstudy-studie/)
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- Diagnose Multiples Myelom,
- Rückfall (Rezidiv) oder Fortschreiten (Progress) des Multiplen Myeloms unter oder nach einer autologen Stammzelltransplantation
- Erhalt von höchstens 1 Zyklus einer sogenannten Salvage-Therapie (Rezidiv-Therapie) vor Einschluss in die Studie
Ausschlusskriterien:
- Unzureichende Organfunktion gemäß der Definition im Prüfplan
- Aktive Hepatitis B oder C oder unkontrollierte HIV-Infektion
- Andere aktive bösartige Erkrankung
- Vorherige Behandlung mit allogenen Stammzellen
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Belantamab-Mafodotin
Eine Phase-III-Studie zu Belantamab-Mafodotin plus Pomalidomid und Dexamethason vs.Pomalidomid, Bortezomib und Dexamethason bei Studienteilnehmern mit rezidiviertem/refraktärem
multiplem Myelom (RRMM)
Organisatorische Daten:
| Prüfplancode: | 207499 |
| ISRCTN: | |
| EudraCT: | 2018-004354-21 |
| Clinicaltrials.gov: | NCT04484623 |
| DRKS: | |
| Sponsor: | GlaxoSmithKline |
| Studienphase: | III |
| Status: | Rekrutierung geplant bis Jani/2027 |
Ziel:
Der primäre Endpunkt der Studie ist das progressionsfreie Überleben. Sekundäre Endpunkte sind u.a. die Erfassung des Überlebens, der Ansprechrate, der Dauer des Ansprechens sowie Aspekte zur Sicherheit und Lebensqualität.
Hintergrund:
Bei der Studie DREAMM-8 handelt es sich um eine offene, randomisierte Phase-III-Studie zur Beurteilung der Wirksamkeit und der Sicherheit von Belantamab-Mafodotin in Kombination mit
Pomalidomid und Dexamethason (B-Pd) im Vergleich zu Pomalidomid, Bortezomib und Dexamethason (PVd) bei Studienteilnehmern mit RRMM, die mit Lenalidomid vorbehandelt wurden
und mindestens 1 vorherige Therapielinie erhalten haben.
Die Studienteilnehmer werden in einem Verhältnis von 1:1 zentral in einen der beiden Behandlungsarme randomisiert. Es ist geplant ca. 450 Patienten in die Studie aufzunehmen. Diese
werden bis zum Eintreten eines der folgenden Ereignisse behandelt: Fortschreiten der Erkrankung, Tod, schwere Nebenwirkungen, Beginn einer neuen Krebsbehandlung, Ausscheiden aus eigenem Wunsch oder Studienende.
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- Der Patient muss eine bestätigte Diagnose des Multiplen Myeloms gemäß den IMWG-Kriterien
haben - Performance-Status der Eastern Cooperative Oncology Group (ECOG - Index zur Abstufung der
Lebensqualität von Patienten mit Krebserkrankungen) von 0 bis 2 - Der Teilnehmer muss zuvor mit mindestens einer vorherigen MM-Therapielinie einschließlich
eines Lenalidomid enthaltenden Behandlungsschemas behandelt worden sein und das
Fortschreiten der Krankheit muss während oder nach seiner letzten Therapie dokumentiert
worden sein - Der Teilnehmer hat sich einer autologen Stammzellentransplantation unterzogen oder gilt als
nicht transplantationsfähig - Adäquate Organsystem-Funktionen, die im Protokoll genau definiert sind
Ausschlußkriterien:
- Aktive Plasmazell-Leukämie zum Zeitpunkt des Screenings. Symptomatische Amyloidose, aktives
POEMS-Syndrom (Polyneuropathie, Organomegalie, Endokrinopathie, monoklonale
Plasmaproliferationsstörung und Hautveränderungen) - Systemische Anti-Myelom-Therapie (einschließlich Chemotherapie und systemischer Steroide)
oder Verwendung eines Prüfpräparats innerhalb von 14 Tagen oder fünf Halbwertszeiten vor
der ersten Dosis des Prüfmedikaments - Vorherige Behandlung mit einem monoklonalen Antikörpermedikament innerhalb von 30 Tagenvor Erhalt der ersten Dosis der Prüfmedikamente.
- Vorherige gezielte BCMA-Therapie
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Belantamab-Mafodotin in Kombination mit Bortezomib + Lenalidomid + Dexamethason
Organisatorische Daten:
| Prüfplancode: | 209664 |
| ISRCTN: | |
| EudraCT: | 2019-003047-30 |
| Clinicaltrials.gov: | NCT04091126 |
| DRKS: | |
| Sponsor: | GlaxoSmithKline |
| Studienphase: | I |
| Status: | Rekrutierung geplant bis 11/2025 |
Ziel:
In dieser randomisierten, offenen Phase-I-Studie werden die Wirksamkeit und Sicherheit von Belantamab-Mafodotin in Kombination mit Bortezomib, Lenalidomid und Dexamethason (VRd) im Vergleich zu VRd allein bei Patienten mit neu neudiagnostiziertem Multiplen Myelom untersucht, die nicht für eine autologe Stammzelltransplantation geeignet sind.
Durchführung:
Die Studie besteht aus zwei Teilen:
In Teil 1 werden die Sicherheit und Verträglichkeit von Belantamab-Mafodotin in bis zu 5 Dosisstufen in Kombination mit VRd bewertet und die empfohlene Dosis für die Phase III (Teil 2) ermittelt.
In Teil 2 (Verlängerung) werden die Wirksamkeit und Sicherheit der für die Phase III empfohlenen Dosis von Belantamab-Mafodotin in Kombination mit VRd im Vergleich zu VRd allein bewertet.
Hintergrund:
Belantamab-Mafodotin (vormals GSK2857916) ist ein gegen das Oberflächenantigen BCMA (engl. B-Cell Maturation Antigen) gerichtetes Antikörper-Wirkstoff-Konjugat. Sobald Belantamab an BCMA bindet, wird das krebstötende Molekül in die Zelle freigesetzt und führt den Zelltod herbei. Bei stark vortherapierten Patienten mit rezidiviertem/refraktärem multiplem Myelom hat Belantamab-Mafodotin eine hohe Wirksamkeit gezeigt. Weitere Untersuchungen müssen zeigen, inwieweit auftretende Nebenwirkungen, teilweise mit vorübergehender Beeinträchtigung des Sehvermögens, die Einsatzmöglichkeiten dieser Therapie einschränken. Vielversprechend erscheinen Kombinationen mit etablierten Substanzen zur weiteren Verbesserung der Ansprechrate, -tiefe, und -dauer.
Belantamab-Mafodotin ist noch nicht zur Anwendung zugelassen. Allerdings wird der Antrag auf Zulassung des Arzneimittels für die Behandlung von Patienten mit rezidiviertem oder therapierefraktärem Multiplen Myelom im Rahmen eines beschleunigten Verfahrens derzeit sowohl von der amerikanischen Zulassungsbehörde FDA als auch der europäischen Arzneimittelbehörde EMA geprüft. (Stand der Information: Februar 2020, https://myelomaresearchnews.com/2020/02/05/ema-review-gsks-belantamab-mafodotin-treatment-heavily-pretreated-multiple-myeloma/
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
-
ECOG Performance Status (Index zur Abstufung der Lebensqualität von Patienten mit Krebserkrankungen) von 0 bis2
-
Neudiagnose Multiples Myelom
-
Der Patient ist für die Hochdosistherapie und die autologe Stammzelltransplantation nicht geeignet
-
Der Patient erfüllt mindestens eines der folgenden Kriterien einer messbaren Krankheitsaktivität:
Urin-Leichtkette (M-Protein) ≥ 200 mg/24 Stunden ≥ 0,2 g/24 Stunden) oder
Serum M-Protein ≥ 0,5 g/l ≥ 5,0 g/l]) oder
Konzentration der betroffenen freien Leichtkette im Serum (sFLC) ≥ 10 mg/dl (≥ 100 mg/l) und abnormes sFLC-Verhältnis (< 0,26 oder > 1,65).
Ausschlußkriterien:
- Smouldering Myelom (SMM)
- Systemische Vortherapie (mit Ausnahme von insgesamt bis zu 160 mg Dexamethason über 4 Tage)
- Periphere Neuropathie ≥ Grad
- Größere Operation innerhalb von 4 Wochen vor der ersten Gabe der Studienmedikation
- Keratopathie
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Induktionstherapie mit Lenalidomid, Bortezomib und Dexamethason und entweder intravenösem oder subkutanem Isatuximab bei Patienten mit neu diagnostiziertem Multiplem Myelom, die für eine Hochdosis-Chemotherapie mit nachfolgender autologer Stammzelltransplantation geeignet sind
Organisatorische Daten:
| Prüfplancode: | GMMG-HD 8 / DSMM XIX |
| ISRCTN: | |
| EudraCT: | 2022-000996-38 |
| Clinicaltrials.gov: | NCT05804032 |
| DRKS: | |
| Sponsor: | Universitätsklinikum Heidelberg DSMM, Sanofi und KKS-Netzwerk |
| Studienphase: | |
| Status: | Rekrutierung geplant bis Juni 2025 |
Ziel:
In dieser Studie für Patienten mit zuvor unbehandeltem Multiplem Myelom soll die Wirksamkeit der Induktionstherapie mit Isatuximab in Kombination mit Lenalidomid/Bortezomib/Dexamethason (RVd) bei Gabe von Isatuximab unter die Haut (subkutan) gegenüber einer Gabe in die Vene (intravenös) untersucht werden.
Durchführung:
Randomisierung: Die Patienten werden vor der Induktionstherapie nach dem Zufallsverfahren (randomisiert) einem von 2 Studienarmen (A oder B) zugeordnet.
- Patienten in Arm A erhalten 3 Zyklen des monoklonalen Antikörpers Isatuximab intravenös in Kombination mit dem RVd-Schema (Lenalidomid, Bortezomib, Dexamethason). Jeder Zyklus hat eine Dauer von 42 Tagen.
- Patienten in Arm B erhalten 3 Zyklen Isatuximab subkutan in Kombination mit RVd.
Nach der Induktionstherapie erhalten die Patienten eine Standard-Intensivierung (in der Regel eine Mobilisierungstherapie auf Cyclophosphamid-Basis, Stammzellenentnahme und hochdosiertes Melphalan, gefolgt von einer autologen Stammzelltransplantation
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- Bestätigte Diagnose eines neu diagnostizierten behandlungsbedürftigen Multiplen Myeloms gemäß den überarbeiteten Diagnosekriterien der International Myeloma Working Group (IMWG))
- Der Patient ist für eine Hochdosis-Chemotherapie und eine autologe Stammzelltransplantation geeignet
- Messbare Krankheitsaktivität (gemäß der Definition im Prüfplan)
- Alter 18 bis 70 Jahre bei Studieneinschluss
Ausschlußkriterien:
- Bekannte Überempfindlichkeit (oder Kontraindikation) gegen einen der Bestandteile der Studientherapie
- Systemische Amyloid-Leichtketten-Amyloidose (außer lokalisierte Amyloid-Leichtketten -Amyloidose, die auf die Haut oder das Knochenmark beschränkt ist)
- Plasmazell-Leukämie
- Vorherige Chemo- oder Strahlentherapie in den letzten 5 Jahren (außer lokaler Strahlentherapie bei lokalem Fortschreiten des Myeloms)
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Erstanwendung von REGN5458 am Menschen bei Patienten mit rezidiviertem oder refraktärem Multiplem Myelom (MM)
Organisatorische Daten:
| Prüfplancode: | R5458-ONC-1826 |
| ISRCTN: | |
| EudraCT: | 2018-003188-78 |
| Clinicaltrials.gov: | NCT03761108 |
| DRKS: | |
| Sponsor: | Regeneron Pharmaceuticals |
| Studienphase: | Phase 1/2 Erstanwendung am Menschen |
| Status: | Rekrutierung läuft, geplant bis Mai 2025 |
Ziele:
Im Phase-1-Teil der Studie: Bewertung der Sicherheit, Verträglichkeit und dosisbegrenzender Nebenwirkungen (dosislimitierender Toxizitäten) und Ermittlung eines für die Phase 2 empfohlenen Dosierungsschemas von REGN5458 als Monotherapie bei Patienten mit rezidiviertem oder refraktärem multiplem Myelom.
Im Phase-2-Teil der Studie: Bewertung der Anti-Tumor-Aktivität von REGN5458, gemessen an der objektiven Ansprechrate und bestimmt durch ein unabhängiges Prüfungskomitee.
Hintergrund:
REGN5458 ist ein Antikörper, der gegen zwei Ziele gerichtet (bispezifisch) ist. Er soll an das Oberflächenprotein BCMA (B-cell maturation antigen) auf multiplen Myelomzellen und an den CD3-Rezeptor auf T-Zellen binden, um sie miteinander zu verbinden und die T-Zellen zur Abtötung der Krebszellen zu aktivieren.
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- ECOG Performance Status (Index zur Abstufung der Lebensqualität von Patienten mit Krebserkrankungen) von 0 oder 1.
- Bestätigte Diagnose eines aktiven Multiplen Myeloms gemäß den von der International Myeloma Working Group (IMWG) aufgestellten Diagnosekriterien
- Das Ansprechen muss anhand der Ansprechkriterien der IMWG auswertbar sein.
-
Phase 1 Dosissteigerung:
- Patienten mit MM, die alle Behandlungsmöglichkeiten ausgeschöpft haben, von denen ein bedeutsamer klinischer Nutzen zu erwarten ist, was sich entweder durch einen Krankheitsrückfall, eine behandlungsrefraktäre Erkrankung oder eine Unverträglichkeit der Therapie zeigt und einen der folgenden Punkte umfasst:
- Fortschreiten der Erkrankung während oder nach mindestens drei Therapielinien oder Unverträglichkeit der Therapie, einschließlich eines Proteasom-Inhibitors, eines immunmodulatorischen Wirkstoffs (IMiD) und eines Anti-CD38-Antikörpers ODER
- Fortschreiten der Krankheit während oder nach einem Anti-CD38-Antikörper und „doppelte Refraktärität“ gegenüber einem Proteasom-Inhibitor und einem IMiD oder Unverträglichkeit der Therapie. Der Anti-CD38-Antikörper kann allein oder in Kombination mit einem anderen Wirkstoff, z. B. einem Proteasom-Inhibitor, verabreicht worden sein. Eine refraktäre Erkrankung ist definiert als Ausbleiben des Ansprechens oder Rückfall innerhalb von 60 Tagen nach der letzten Behandlung.
Phase 2:
- Die Patienten müssen dreifach refraktär sein, d. h. sie müssen auf eine vorherige Behandlung mit mindestens einem Anti-CD38-Antikörper, einem Proteasom-Inhibitor und einem IMiD refraktär sein. Darüber hinaus müssen die Patienten fünf Wirkstoffe erhalten haben (d. h. sie müssen zuvor mit zwei Proteasom-Inhibitoren, zwei IMiDs [Lenalidomid und Pomalidomid] und einem monoklonalen Anti-CD38-Antikörper behandelt worden sein).
Ausschlusskriterien:
- Diagnose einer Plasmazellleukämie, einer primären systemischen Leichtketten-Amyloidose (ausgenommen Myelom-assoziierte Amyloidose), einer Waldenström-Makroglobulinämie (lymphoplasmatisches Lymphom) oder eines POEMS-Syndroms (Polyneuropathie, Organomegalie, Endokrinopathie, monoklonale Proteine und Hautveränderungen)
- Patienten mit bekannten MM-Hirnläsionen oder meningealer Beteiligung
- Vorherige Behandlung mit BCMA-gerichteten Immuntherapien, einschließlich BCMA-bispezifischer Antikörper und BiTEs sowie BCMA-CAR-T-Zellen.
- Hinweis: BCMA-Antikörper-Wirkstoff-Konjugate sind nicht ausgeschlossen
- Vorgeschichte einer allogenen Stammzelltransplantation zu irgendeinem Zeitpunkt oder einer autologen Stammzelltransplantation innerhalb von 12 Wochen vor Beginn der Studienbehandlung
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt
Speyer
Onkologische Schwerpunktpraxis Speyer
06232- 60 44 60
Studien
GMMG-HD8/DSMM-XIX
Induktionstherapie mit Lenalidomid, Bortezomib und Dexamethason und entweder intravenösem oder subkutanem Isatuximab bei Patienten mit neu diagnostiziertem Multiplem Myelom, die für eine Hochdosis-Chemotherapie mit nachfolgender autologer Stammzelltransplantation geeignet sind
Organisatorische Daten:
| Prüfplancode: | GMMG-HD 8 / DSMM XIX |
| ISRCTN: | |
| EudraCT: | 2022-000996-38 |
| Clinicaltrials.gov: | NCT05804032 |
| DRKS: | |
| Sponsor: | Universitätsklinikum Heidelberg DSMM, Sanofi und KKS-Netzwerk |
| Studienphase: | |
| Status: | Rekrutierung geplant bis Juni 2025 |
Ziel:
In dieser Studie für Patienten mit zuvor unbehandeltem Multiplem Myelom soll die Wirksamkeit der Induktionstherapie mit Isatuximab in Kombination mit Lenalidomid/Bortezomib/Dexamethason (RVd) bei Gabe von Isatuximab unter die Haut (subkutan) gegenüber einer Gabe in die Vene (intravenös) untersucht werden.
Durchführung:
Randomisierung: Die Patienten werden vor der Induktionstherapie nach dem Zufallsverfahren (randomisiert) einem von 2 Studienarmen (A oder B) zugeordnet.
- Patienten in Arm A erhalten 3 Zyklen des monoklonalen Antikörpers Isatuximab intravenös in Kombination mit dem RVd-Schema (Lenalidomid, Bortezomib, Dexamethason). Jeder Zyklus hat eine Dauer von 42 Tagen.
- Patienten in Arm B erhalten 3 Zyklen Isatuximab subkutan in Kombination mit RVd.
Nach der Induktionstherapie erhalten die Patienten eine Standard-Intensivierung (in der Regel eine Mobilisierungstherapie auf Cyclophosphamid-Basis, Stammzellenentnahme und hochdosiertes Melphalan, gefolgt von einer autologen Stammzelltransplantation
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- Bestätigte Diagnose eines neu diagnostizierten behandlungsbedürftigen Multiplen Myeloms gemäß den überarbeiteten Diagnosekriterien der International Myeloma Working Group (IMWG))
- Der Patient ist für eine Hochdosis-Chemotherapie und eine autologe Stammzelltransplantation geeignet
- Messbare Krankheitsaktivität (gemäß der Definition im Prüfplan)
- Alter 18 bis 70 Jahre bei Studieneinschluss
Ausschlußkriterien:
- Bekannte Überempfindlichkeit (oder Kontraindikation) gegen einen der Bestandteile der Studientherapie
- Systemische Amyloid-Leichtketten-Amyloidose (außer lokalisierte Amyloid-Leichtketten -Amyloidose, die auf die Haut oder das Knochenmark beschränkt ist)
- Plasmazell-Leukämie
- Vorherige Chemo- oder Strahlentherapie in den letzten 5 Jahren (außer lokaler Strahlentherapie bei lokalem Fortschreiten des Myeloms)
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Trier
Klinikum Mutterhaus der Borromäerinnen, Akademisches Lehrkrankenhaus der Johannes Gutenberg-Universität Mainz
0651-947-0
Unterkategorien
-
Kaiserslautern
- COM_STUDIES_NUM
- 1
-
Koblenz
- COM_STUDIES_NUM
- 3
-
Ludwigshafen
- COM_STUDIES_NUM
- 1
-
Mainz
- COM_STUDIES_NUM
- 1
-
Speyer
- COM_STUDIES_NUM
- 1
-
Trier
- COM_STUDIES_NUM
- 1
