COM_STUDIES_DEFAULT_PAGE_TITLE
Hamburg
Hamburg
Asklepiosklinik Altona, Hämatologie und internistische Onkologie
040-181881-1273
Studien
AlloRelapseMM
Allogene Stammzelltransplantation im Vergleich zur konventionellen Therapie als Salvage-Therapie für Patienten mit rezidiviertem / progredientem Multiplen Myelom nach einer Erstlinientherapie (AlloRelapseMMStudy)
Organisatorische Daten:
| Prüfplancode: | |
| ISRCTN: | |
| EudraCT: | 2021-001--5-67 |
| Clinicaltrials.gov: | NCT0567319 |
| DRKS: | |
| Sponsor: | Universitätsklinikum Hamburg-Eppendorf mit Unterstützung des Gemeinsamen Bundesausschusses (G-BA) |
| Studienphase: | III |
| Status: | Rekrutierung läuft, geplant bis 6/2027 |
Ziel:
In dieser Studie wird untersucht, ob die Transplantation von gespendeten Stammzellen (allogene Stammzelltransplantation) Vorteile gegenüber der gängigen Therapie bei Patienten mit Multiplem Myelom bietet, bei denen es nach einer Transplantation von körpereigenen Stammzellen (autologe Stammzelltransplantation) als Erstlinientherapie zu einem Erkrankungsrückfall (Rezidiv) oder einem Fortschreiten der Krankheit (Progression) gekommen ist. Es wird geprüft, ob die allogene Stammzelltransplantation das Gesamtüberleben bei diesen Patienten verlängert.
Hintergrund:
Eine Behandlungsoption des Multiplen Myeloms ist die Stammzelltransplantation. Nach einer Chemotherapie, bei der die Krebszellen zerstört werden, werden körpereigene (autologe) oder gespendete körperfremde (allogene) Stammzellen übertragen. Die übertragenen Stammzellen siedeln sich dann im Knochenmark an und bilden „frische“ Blutzellen. Die Stammzelltransplantation kommt als Behandlungsoption nach der ersten Diagnose (Erstlinientherapie), aber auch bei einem Rückfall nach Vorbehandlung (jenseits der Erstlinientherapie) in Frage. Auch wenn das Multiple Myelom oft nicht endgültig heilbar ist, soll die Therapie die Lebenserwartung verlängern und die Symptome zurückdrängen. (Quelle: https://www.g-ba.de/studien/erprobung/allorelapsemmstudy-studie/)
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- Diagnose Multiples Myelom,
- Rückfall (Rezidiv) oder Fortschreiten (Progress) des Multiplen Myeloms unter oder nach einer autologen Stammzelltransplantation
- Erhalt von höchstens 1 Zyklus einer sogenannten Salvage-Therapie (Rezidiv-Therapie) vor Einschluss in die Studie
Ausschlusskriterien:
- Unzureichende Organfunktion gemäß der Definition im Prüfplan
- Aktive Hepatitis B oder C oder unkontrollierte HIV-Infektion
- Andere aktive bösartige Erkrankung
- Vorherige Behandlung mit allogenen Stammzellen
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Studie zur Untersuchung alternativer Dosierungsschemata von Belantamab-Mafodotin bei Teilnehmern mit rezidiviertem oder refraktärem
Multiplem Myelom (RRMM) (DREAMM 14)
Organisatorische Daten:
| Prüfplancode: | 209628 |
| ISRCTN: | |
| EudraCT: | 2021-004151-16 |
| Clinicaltrials.gov: | NCT05064358 |
| DRKS: | |
| Sponsor: | GlaxoSmithKline |
| Studienphase: | 2 |
| Status: | Rekrutierung geplant bis Mai 2024 |
Ziel:
Ziel dieser Studie ist es, alternative Dosierungsschemata von Belantamab-Mafodotin als Einzelwirkstoff bei Teilnehmern mit rezidiviertem oder refraktärem Multiplem Myelom (RRMM) zu untersuchen. So soll festgestellt werden, ob ein verbessertes Nutzen-/Risikoprofil insgesamt erreicht werden kann, wenn die Belantamab-Mafodotin-Dosis, der Zeitplan der Verabreichung oder beides geändert werden.
Hintergrund:
Belantamab-Mafodotin ist ein Antikörper-Wirkstoff-Konjugat, das zur Behandlung von Krebserkrankungen eingesetzt wird. Es ist richtet sich gegen das B-Zell-Reifungsantigen (BCMA).
BCMA ist auf der Oberfläche CD138-positiver Myelomzellen nachweisbar, nicht jedoch auf gesunden B-Zellen. Daher bietet sich BCMA als Zielstruktur in der Myelomtherapie an. Belantamab-Mafodotin ist für die Behandlung des Multiplen Myeloms zugelassen.
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- ECOG Performance Status (Index zur Abstufung der Lebensqualität von Patienten mit Krebserkrankungen) von 0 bis 2
- Der Patient hat eine histologisch oder zytologisch bestätigte Diagnose eines MM und
- a) hat sich zuvor einer Stammzelltransplantation unterzogen oder gilt als nicht transplantationsfähig, und
- b) hat auf mindestens drei vorherige Therapielinien für MM, einschließlich eines CD38-Antikörpers (z. B. Daratumumab) allein oder in Kombination, nicht angesprochen. Zudem ist der Patient refraktär gegenüber einem immunmodulatorischen Mittel (z. B. Lenalidomid, Pomalidomid) und einem Proteasom-Inhibitor (z. B. Bortezomib, Ixazomib, Carfilzomib).
- Der Teilnehmer hat eine messbare Erkrankung gemäß den modifizierten IMWG-Kriterien.
Ausschlußkriterien:
- Symptomatische Amyloidose, aktives POEMS-Syndrom (Polyneuropathie, Organomegalie, Endokrinopathie, Myelom-Protein und Hautveränderungen) oder aktive Plasmazellleukämie zum Zeitpunkt des Sreenings.
- Aktuelle Erkrankung des Hornhautepithels, ausgenommen nicht-konfluente, oberflächliche, punktförmige Keratitis (SPK).
- Anzeichen für aktive Schleimhaut- oder innere Blutungen.
- aktive Nierenerkrankung.
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Teclistamab, Daratumumab, Lenalidomid,Dexamethason +/- Bortezomib
Phase-2-Studie zur Bewertung der Sicherheit und Wirksamkeit von Teclistamab in Kombination mit Daratumumab, Lenalidomid und Dexamethason mit und ohne Bortezomib als Induktionstherapie und Teclistamab in Kombination mit Daratumumab und Lenalidomid als Erhaltungstherapie bei transplantationsgeeigneten Patienten mit neu diagnostiziertem Multiplem Myelom
Organisatorische Daten:
| Prüfplancode: | GMMG-HD10, DSMM-XX, 64007957MMY2003, MajesTEC-5 |
| ISRCTN: | |
| EudraCT: | |
| Clinicaltrials.gov: | NCT05695508 |
| DRKS: | |
| Sponsor: | Universitätsklinikum Heidelberg in Zusammenarbeit mit Janssen |
| Studienphase: | Phase 3 |
| Status: | Rekrutierung läuft, geplant bis Mai 2026 |
Hintergrund:
Teclistamab ist ein bispezifischer Antikörper, der an das B-Zell-Reifungsantigen (BCMA), ein Protein auf der Oberfläche von Myelomzellen, bindet und gegen CD3-Rezeptoren auf der Oberfläche von T-Zellen gerichtet ist. Teclistamab wurde im August 2022 in der EU als Monotherapie zur Behandlung des fortgeschrittenen Multiplen Myeloms zugelassen und wird jetzt bei weiteren Indikationen geprüft.
In dieser Studie bei Patienten mit neu diagnostiziertem Multiplem Myelom, die für eine Transplantation geeignet sind, werden die Sicherheit, Verträglichkeit und Wirksamkeit einer Fünffach-Kombination in der Induktionstherapie geprüft: Teclistamab (Tec) in Kombination mit Daratumumab (Dara), Lenalidomid (R), Bortezomib (V) und Dexamethason (d).
Durchführung
Das Studiendesign gliedert sich in drei Studienarme:
Patienten in Arm A und B erhalten eine Induktionstherapie mit Tec-Dara-Rd (Arm A) oder Tec-Dara-VRd (Arm B). Es folgen eine Hochdosis-Chemotherapie und eine autologe Stammzelltransplantation gemäß Therapiestandard sowie eine Erhaltungstherapie mit Tec-Dara-R.
Patienten in Arm C erhalten eine Induktionstherapie gemäß Therapiestandard gefolgt von einer Hochdosis-Chemotherapie und einer autologen Stammzelltransplantation sowie gegebenenfalls einer Konsolidierungstherapie. Im Anschluss daran folgt eine Erhaltungstherapie mit Tec-Dara-R.
Die Behandlungsdauer der Induktionsphase beträgt 6 Zyklen (à 28 Tage). Im Anschluss daran folgen eine Hochdosis-Chemotherapie und autologe Stammzelltransplantation . Danach beginnt die Erhaltungsphase der Studie unter Tec-Dara-R über 18 Zyklen.
Studienarm C beginnt mit dem Screening nach abgeschlossener Hochdosis-Chemotherapieplus autologer Stammzelltransplantation. Die Behandlung erfolgt genauso wie in Arm A und B mit Tec-Dara-R als Erhaltungstherapie für 18 Zyklen.
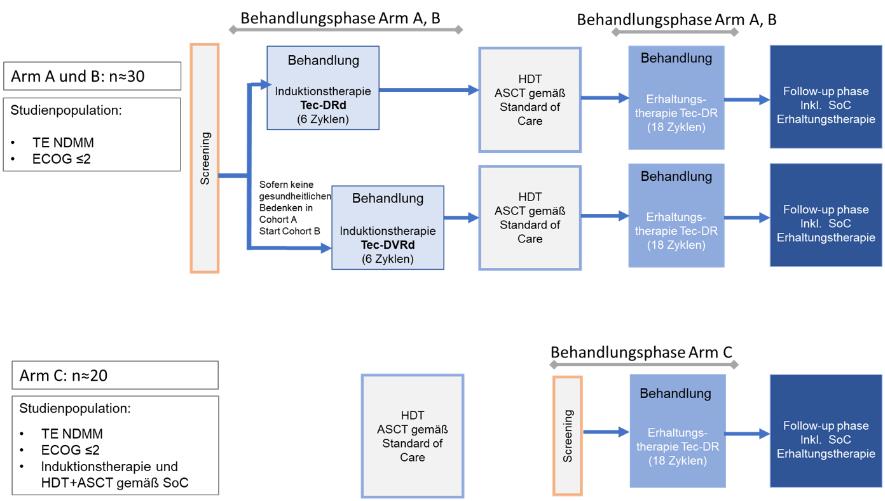
Abb. Studiendesign, mit freundlicher Genehmigung des Universitätsklinikums Heidelberg, Prof. Raab
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- Neu diagnostiziertes Multiples Myelom gemäß den Diagnosekriterien der International Myeloma Working Group (IMWG) und Erhalt einer Induktionstherapie mit oder ohne Konsolidierung
- Ein ECOG-Performance-Status-Score (Index zur Abstufung der Lebensqualität von Patienten mit Krebserkrankungen) von 0, 1 oder 2 beim Screening und unmittelbar vor Beginn der Verabreichung der Studienbehandlung
- Messbare Erkrankung während der Screening-Phase, gemäß der Definition im Protokoll
Ausschlußkriterien
- Beteiligung des ZNS oder klinische Anzeichen für eine Beteiligung der Hirnhäute
- Schlaganfall oder Krampfanfall innerhalb von 6 Monaten vor Studienbeginn
- Transplantation in der Krankengeschichte, die eine immunsuppressive Behandlung erforderte
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Induktionstherapie mit Lenalidomid, Bortezomib und Dexamethason und entweder intravenösem oder subkutanem Isatuximab bei Patienten mit neu diagnostiziertem Multiplem Myelom, die für eine Hochdosis-Chemotherapie mit nachfolgender autologer Stammzelltransplantation geeignet sind
Organisatorische Daten:
| Prüfplancode: | GMMG-HD 8 / DSMM XIX |
| ISRCTN: | |
| EudraCT: | 2022-000996-38 |
| Clinicaltrials.gov: | NCT05804032 |
| DRKS: | |
| Sponsor: | Universitätsklinikum Heidelberg DSMM, Sanofi und KKS-Netzwerk |
| Studienphase: | |
| Status: | Rekrutierung geplant bis Juni 2025 |
Ziel:
In dieser Studie für Patienten mit zuvor unbehandeltem Multiplem Myelom soll die Wirksamkeit der Induktionstherapie mit Isatuximab in Kombination mit Lenalidomid/Bortezomib/Dexamethason (RVd) bei Gabe von Isatuximab unter die Haut (subkutan) gegenüber einer Gabe in die Vene (intravenös) untersucht werden.
Durchführung:
Randomisierung: Die Patienten werden vor der Induktionstherapie nach dem Zufallsverfahren (randomisiert) einem von 2 Studienarmen (A oder B) zugeordnet.
- Patienten in Arm A erhalten 3 Zyklen des monoklonalen Antikörpers Isatuximab intravenös in Kombination mit dem RVd-Schema (Lenalidomid, Bortezomib, Dexamethason). Jeder Zyklus hat eine Dauer von 42 Tagen.
- Patienten in Arm B erhalten 3 Zyklen Isatuximab subkutan in Kombination mit RVd.
Nach der Induktionstherapie erhalten die Patienten eine Standard-Intensivierung (in der Regel eine Mobilisierungstherapie auf Cyclophosphamid-Basis, Stammzellenentnahme und hochdosiertes Melphalan, gefolgt von einer autologen Stammzelltransplantation
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- Bestätigte Diagnose eines neu diagnostizierten behandlungsbedürftigen Multiplen Myeloms gemäß den überarbeiteten Diagnosekriterien der International Myeloma Working Group (IMWG))
- Der Patient ist für eine Hochdosis-Chemotherapie und eine autologe Stammzelltransplantation geeignet
- Messbare Krankheitsaktivität (gemäß der Definition im Prüfplan)
- Alter 18 bis 70 Jahre bei Studieneinschluss
Ausschlußkriterien:
- Bekannte Überempfindlichkeit (oder Kontraindikation) gegen einen der Bestandteile der Studientherapie
- Systemische Amyloid-Leichtketten-Amyloidose (außer lokalisierte Amyloid-Leichtketten -Amyloidose, die auf die Haut oder das Knochenmark beschränkt ist)
- Plasmazell-Leukämie
- Vorherige Chemo- oder Strahlentherapie in den letzten 5 Jahren (außer lokaler Strahlentherapie bei lokalem Fortschreiten des Myeloms)
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Studie zur Bewertung der Sicherheit von HDP-101 bei Patienten mit rezidiviertem refraktärem multiplem Myelom
Organisatorische Daten:
| Prüfplancode: | HDP-101-01 |
| ISRCTN: | |
| EudraCT: | 2020-003414-12 |
| Clinicaltrials.gov: | NCT04879043 |
| DRKS: | |
| Sponsor: | Heidelberg Pharma |
| Studienphase: | Phase 1/2a Erstanwendung am Menschen |
| Status: | Rekrutierung läuft, geplant bis Mai 2024 |
Hintergrund:
BCMA (B-cell maturation antigen) ist ein Oberflächenprotein, das beim Multiplen Myelom hoch exprimiert wird und an das die ausgewählten Antikörper spezifisch binden. Wissenschaftler des Max Delbrücks Institutes haben diese BCMA-spezifischen Antikörper entwickelt und Heidelberg Pharma hat auf Basis ihrer ATAC-Technologie daraus mehrere proprietäre ATAC-Moleküle hergestellt, mit denen sie umfangreiche präklinische Daten gewinnen konnte. Auf Grundlage dieser Daten wurde der Entwicklungskandidat HDP-101, der sich aus dem BCMA-Antikörper, einem Linker und dem Toxin Amanitin zusammensetzt, für die spätere klinische Entwicklung ausgewählt.
(Quelle: www.heidelberg-pharma.de)
Die Dosiserweiterungsphase der Studie zielt dann darauf ab, erste Hinweise auf eine Antitumoraktivität zu sammeln und die Sicherheit von HDP-101 als Monotherapie zu bestätigen.
Durchführung:
Die Studie besteht aus zwei Teilen: einer Dosis-Steigerungsphase in Teil 1 und einer Expansionsphase in Teil 2a, in der Sicherheit, Verträglichkeit, Pharmakokinetik (PK, Einwirkung des Körpers auf die Substanz), Pharmakodynamik (PD, Einwirkung der Substanz auf den Körper) und klinische Aktivität getestet werden. In die Studie werden Patienten mit rezidiviertem/refraktärem Multiplen Myelom oder anderen Plasmazellerkrankungen, die BCMA exprimieren, aufgenommen.
Die Teilnehmer erhalten eine Dosis HDP-101 intravenös alle drei Wochen (21-Tage-Zyklus) bis zum Fortschreiten der Erkrankung, dem Auftreten einer unverträglichen Nebenwirkung, nach Ermessen des Prüfarztes oder bis zum Ausscheiden des Patienten.
Während der Phase 1 wird die Verträglichkeit der verschiedenen Dosisstufen untersucht. Während des Phase-2a-Dosissteigerungsteils wird die empfohlene Phase-2-Dosis (RP2D) von HDP-101 verabreicht.
Einschlusskriterien:
- Bestätigte Diagnose eines aktiven Multiplen Myeloms gemäß den von der International Myeloma Working Group (IMWG) aufgestellten Diagnosekriterien
- Patient hat Stammzelltransplantation (SZT) erhalten oder gilt als nicht transplantationsfähig
- Der Patient muss zuvor mit einer Antimyelom-Therapie behandelt worden sein, die ein immunmodulatorisches Medikament, einen Proteasom-Inhibitor und eine Anti-CD38-Behandlung - allein oder in Kombination - umfasst. Darüber hinaus sollte der Patient entweder refraktär oder intolerant gegenüber einer etablierten Standardtherapie sein, die dem Patienten nach Einschätzung des Prüfarztes einen sinnvollen klinischen Nutzen bietet.
- Messbare Erkrankung gemäß den IMWG-Kriterien
- ECOG Performance Status von 0 oder 1 (Index zur Abstufung der Lebensqualität von Patienten mit Krebserkrankungen)
Ausschlusskriterien:
- Nur für Patienten, die am Phase-2a-Teil teilnehmen: Frühere Behandlungen mit zugelassenen oder experimentellen gegen BCMA-gerichteten Methoden sind nicht zulässig.
- Bekannte Beteiligung des zentralen Nervensystems
- Plasmazell-Leukämie
- Herzinsuffizienz in der Vorgeschichte
- Autologe oder allogene SZT innerhalb von 12 Wochen vor der ersten Infusion oder Planung einer autologen SZT
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt
Dies ist eine randomisierte, multizentrische, offene Phase-3-Studie zur Bewertung der subkutanen (SC) gegenüber der intravenösen (IV) Verabreichung von Isatuximab in Kombination mit Pomalidomid und Dexamethason (Pd) bei Patienten mit rezidivierendem refraktärem Multiplen Myelom (RRMM), die mindestens eine vorherige Therapielinie mit Lenalidomid und einem Proteasominhibitor (PI) erhalten haben.
Organisatorische Daten:
| Prüfplancode: | EFC15951 |
| ISRCTN: | |
| EudraCT: | 2021-002485-41 |
| Clinicaltrials.gov: | NCT05405166 |
| DRKS: | |
| Sponsor: | Sanofi |
| Studienphase: | Phase 3 |
| Status: | Rekrutierung läuft, geplant bis Mai 2024 |
Durchführung:
Die in Frage kommenden Teilnehmer werden im Verhältnis 1:1 nach dem Zufallsprinzip (randomisiert) einem von 2 Studienarmen zugeordnet:
Arm SC: Isatuximab SC + Pd
Arm IV: Isatuximab IV + Pd
Die Teilnehmer dürfen die Therapie so lange fortsetzen, bis die Krankheit fortschreitet, inakzeptable unerwünschte Ereignisse auftreten, der Teilnehmer den Abbruch der Therapie wünscht oder ein anderer Grund vorliegt, je nachdem, was zuerst eintritt.
Hintergrund:
Isatuximab ist ein gegen Tumoren gerichteter Wirkstoff und besitzt eine abtötende (zytotoxische) Wirkung auf die Myelom-Zellen. Es gehört zur Gruppe der monoklonalen Antikörper für die Behandlung des Multiplen Myeloms bei Erwachsenen. Die Wirkung von Isatuximab beruht auf der Bindung an den CD38-Rezeptor, was zum Zelltod führt.
Isatuximab wird ist als Therapie des rezidivierenden und refraktären Multiplen Myeloms bei Patienten angewendet, die bereits wegen ihres Multiplen Myeloms behandelt wurden. Es wird gemeinsam mit zwei verschiedenen Arzneimittelkombinationen angewendet:
- Pomalidomid und Dexamethason oder
- Carfilzomib und Dexamethason.
Bisher wird Isatuximab als Tropfinfusion in eine Vene verabreicht (intravenöse Infusion). In dieser Studie wird Isatuximab als Injektion unter die Haut (subkutan) geprüft
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien
- Teilnehmer mit Multiplem Myelom, die mindestens eine vorherige Therapielinie mit Lenalidomid und einem Proteasom-Inhibitor erhalten haben
- Teilnehmer mit messbarer Erkrankung definiert als
- M-Protein im Serum (≥ 0,5 g/dl) und/oder
- M-Protein im Urin (≥ 200 mg/24 Stunden) und/oder
- freie Leichtketten (FLC) im Serum (≥10 mg/dl und anormales Serum-FLC-Verhältnis
Ausschlußkriterien:
- Teilnehmer unter 18 Jahren
- Teilnehmer mit primär refraktärem Multiplem Myelom
- Teilnehmer, die nicht auf Anti-CD38 ansprechen bei einer Auswaschphase von weniger als 9 Monaten, oder die monoklonale Anti-CD38-Antikörper nicht vertragen
- Teilnehmer, die vorher mit Pomalidomid behandelt wurden
- Teilnehmer mit einem ECOG (Index zur Abstufung der Lebensqualität von Patienten mit Krebserkrankungen) > 2
- Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Eine Studie zum Vergleich von Teclistamab-Monotherapie mit Pomalidomid, Bortezomib, Dexamethason (PVd) oder Carfilzomib, Dexamethason (Kd) bei Teilnehmern mit rezidiviertem oder refraktärem Multiplem Myelom (MajesTEC-9)
Organisatorische Daten:
| Prüfplancode: | CR109244, 64007957MMY3006 |
| ISRCTN: | |
| EudraCT: | 2022-000928-37 |
| Clinicaltrials.gov: | NCT05572515 |
| DRKS: | |
| Sponsor: | Janssen |
| Studienphase: | Phase 3 |
| Status: | Rekrutierung läuft, geplant bis August 2025 |
Ziele:
Ziel dieser Studie ist es, die Wirksamkeit von Teclistamab mit PVd bzw. Kd bei Patienten mit rezidiviertem oder refraktärem Multiplem Myelom zu vergleichen, die bereits 1 bis 3 Therapielinien einschließlich einen monoklonalen Anti-CD38-Antikörpers und Lenalidomid erhalten haben.
Hintergrund:
Teclistamab ist ein bispezifischer Antikörper, der sowohl gegen das B-Zell-Reifungsantigen (BCMA), ein Protein auf Myelomzellen, als auch gegen CD3-Rezeptoren auf der T-Zell-Oberfläche gerichtet ist. Teclistamab wurde im August 2022 in der EU als Monotherapie zur Behandlung des fortgeschrittenen Multiplen Myeloms zugelassen und wird jetzt bei weiteren Indikationen geprüft.
In der MajesTEC-1-Studie bei stark vorbehandelten Patienten mit Multiplem Myelom (n=165) bewirkte Teclistamab ein hohes und dauerhaftes Ansprechen. Bei einer medianen Nachbeobachtungszeit von etwa 14 Monaten betrug die Gesamtansprechrate 63 Prozent, wobei 39,4 Prozent ein komplettes Ansprechen oder besser erreichten. Diese Daten wurden auf der Jahrestagung 2022 der American Society of Clinical Oncology (ASCO) veröffentlicht.
Durchführung:
Die Studie umfasst eine Screeningphase, eine Behandlungsphase und eine Nachbeobachtungsphase. Die Sicherheit wird durch körperliche Untersuchungen, neurologische Untersuchungen, den ECOG-Leistungsstatus (Eastern Cooperative Oncology Group), klinische Labortests, Vitalparameter und die Überwachung von Nebenwirkungen beurteilt. Die Gesamtdauer der Studie wird bis zu 9 Jahre betragen.
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- Dokumentierte Diagnose eines multiplen Myeloms gemäß den nachstehenden Kriterien: (a) Diagnose des Multiplen Myeloms gemäß den Diagnosekriterien der International Myeloma Working Group (IMWG) (b) Messbare Erkrankung zum Zeitpunkt des Screenings, gemäß der Definition im Protokoll. 1 bis 3 vorangegangene Therapielinien gegen das Myelom, einschließlich mindestens 2 aufeinanderfolgender Zyklen eines monoklonalen Antikörpers gegen Cluster of Differentiation 38 (CD38) in der zugelassenen Dosierung in einer beliebigen vorangegangenen Therapielinie und 2 aufeinanderfolgende Zyklen von Lenalidomid in einer beliebigen vorangegangenen Therapielinie.
- Dokumentierte Anzeichen für ein Fortschreiten der Krankheit oder das Ausbleiben eines Ansprechens auf die letzte Therapielinie, basierend auf der Bestimmung des Ansprechens durch den Prüfarzt nach den Kriterien der IMWG.
- ECOG Performance Status (Index zur Abstufung der Lebensqualität von Patienten mit Krebserkrankungen) von 0 bis 2.
Ausschlusskriterien:
- Eine vorherige gegen das B-Zell-Reifungsantigen (BCMA)-gerichtete Therapie
- Teilnehmer, für den PVd als Kontrolltherapie aus im Protokoll definierten Gründen nicht infrage kommt.
- Teilnehmer, für den Kd als Kontrolltherapie aus im Protokoll definierten Gründen nicht infrage kommt.
- Beteiligung des zentralen Nervensystems (ZNS) oder klinische Anzeichen einer meningealen Beteiligung des Multiplen Myeloms.
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt
Talquetamab
Talquetamab in Kombination mit Daratumumab oder in Kombination mit Daratumumab und Pomalidomid im Vergleich zu Daratumumab in Kombination mit Pomalidomid und Dexamethason bei Teilnehmern mit rezidiviertem oder refraktärem Multiplem Myelom (MonumenTAL-3)
Organisatorische Daten:
| Prüfplancode: | CR109082, 64407564MMY3002 |
| ISRCTN: | |
| EudraCT: | 2021-000202-22 |
| Clinicaltrials.gov: | NCT05455320 |
| DRKS: | |
| Sponsor: | Janssen |
| Studienphase: | 3 |
| Status: | Rekrutierung geplant bis Febr 2029 |
Ziel:
Ziel der Studie ist der Vergleich der Wirksamkeit von unter die Haut (subkutan, s.c.) verabreichtem Talquetamab in Kombination mit Daratumumab s.c. und Pomalidomid (Tal-DP) bzw. Talquetamab s.c. in Kombination mit Daratumumab s.c. (Tal-D) gegenüber Daratumumab s.c. in Kombination mit Pomalidomid und Dexamethason (DPd) bei Teilnehmern mit rezidiviertem oder refraktärem Multiplem Myelom, die mindestens eine vorherige Therapielinie erhalten haben.
Hintergrund:
Talquetamab ist ein humanisierter monoklonaler Antikörper, der gegen zwei Ziele gerichtet (bispezifisch) ist: gegen humanes CD3, ein Oberflächenantigen auf T-Zellen, und gegen GPRC5D (G protein-coupled receptor family C group 5 member D), ein Antigen in Tumorzellen oder auf deren Oberfläche (tumor-assoziiertes Antigen). Bei Verabreichung bindet Talquetamab sowohl an CD3 auf T-Zellen als auch an GPRC5D, das auf bestimmten Tumorzellen wie z.B. dem Multiplen Myelom vermehrt vorkommt und eine Schlüsselrolle bei der Vermehrung von Tumorzellen spielt.
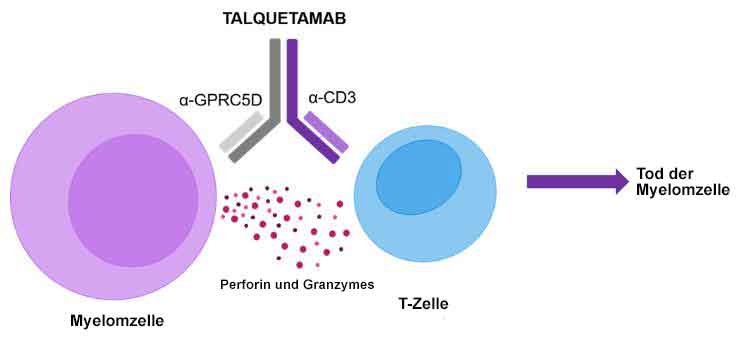
Abb. aus https://multiplemyelomahub.com/,
Durchführung:
Die Studie ist in drei Phasen unterteilt: Voruntersuchung (Screening), Behandlung (bis eines der folgenden Ereignisse eintritt: bestätigtes Fortschreiten der Krankheit, Tod, unverträgliche Toxizität, Widerruf der Einwilligung oder Ende der Studie) und Nachbeobachtung (bis eines der folgenden Ereignisse eintritt: Tod, Widerruf der Einwilligung, Verlust der Nachbeobachtung oder Ende der Studie). Wirksamkeit, Sicherheit, Pharmakokinetik, Immunogenität und Biomarker werden zu bestimmten Zeitpunkten untersucht. Die Gesamtdauer der Studie beträgt bis zu 6 Jahren und 6 Monaten.
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- Dokumentiertes Multiples Myelom, definiert als
- a) Diagnose des Multiplen Myeloms gemäß den diagnostischen Kriterien der International Myeloma Working Group (IMWG) und
- b) messbare Erkrankung zum Zeitpunkt des Screenings gemäß der Definition im Protokoll
- Rezidivierte oder refraktäre Erkrankung gemäß folgender Definition:
- eine rezidivierte Erkrankung ist definiert als anfängliches Ansprechen auf die vorherige Behandlung, gefolgt von einer nach IMWG-Kriterien bestätigten fortschreitenden (progredienten) Erkrankung mehr als 60 Tage nach Beendigung der Behandlung;
- eine refraktäre Erkrankung ist definiert als eine Verringerung des monoklonalen Paraproteins (M-Protein) um weniger als 25 % oder eine nach IMWG-Kriterien bestätigte fortschreitende Erkrankung während der vorherigen Behandlung oder ≤ 60 Tage nach Beendigung der Behandlung
- Teilnehmer müssen mindestens eine vorherige Linie einer Myelom-Therapie, einschließlich eines Proteasom-Inhibitors (PI) und Lenalidomid, erhalten haben. Patienten, die nur eine vorherige Linie einer Myelom-Therapie erhalten haben, müssen als Lenalidomid-refraktär gelten (d. h. bei ihnen ist bei Beendigung einer Lenalidomid-haltigen Therapie oder innerhalb von 60 Tagen danach eine fortschreitende (progrediente) Erkrankung nach IMWG-Kriterien nachweisbar). Teilnehmer, die 2 oder mehr vorherige Therapielinien zur Behandlung des Myeloms erhalten haben, müssen als Lenalidomid-behandelt gelten
- Dokumentierte Anzeichen einer fortschreitenden Erkrankung, basierend auf der Feststellung des Ansprechens durch den Prüfarzt gemäß den IMWG-Kriterien während oder nach der letzten Behandlung
- ECOG (Index zur Abstufung der Lebensqualität von Patienten mit Krebserkrankungen) von 0, 1 oder 2
Ausschlußkriterien
- Gegenanzeigen oder lebensbedrohliche Allergien, Überempfindlichkeiten oder Unverträglichkeiten gegenüber Hilfsstoffen des Studienmedikaments
- Refraktär gegenüber einem monoklonalen Antikörper gegen Cluster of Differentiation 38 (CD38), wie in den IMWG-Konsensrichtlinien definiert (Fortschreiten der Erkrankung während der Behandlung oder innerhalb von 60 Tagen nach Beendigung der Therapie mit einem monoklonalen Antikörper gegen CD38)
- Erhalt von Kortikosteroiden in einer maximalen Gesamtdosis von oder entsprechend ≥ 140 mg Prednison innerhalb von 14 Tagen vor Gabe der ersten Dosis des Studienmedikaments
- Bekannte aktive Beteiligung des zentralen Nervensystems (ZNS) oder klinische Anzeichen einer meningealen Beteiligung des Multiplen Myeloms. Bei Verdacht auf eine der beiden Erkrankungen sind eine negative Magnetresonanztomographie (MRT) des gesamten Gehirns und eine lumbale Zytologie erforderlich.
- Plasmazellleukämie zum Zeitpunkt der Voruntersuchung, Waldenström-Makroglobulinämie, Polyneuropathie, Organomegalie, Endokrinopathie, M-Protein und Hautveränderungen (POEMS-Syndrom) oder primäre Amyloid-Leichtketten-Amyloidose
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Asklepiosklinik St. Georg
040-181885-2005
Studien
EXCALIBER-RRMMPhase 3- Studie zu Iberdomid
Offene Studie zum Vergleich von Iberdomid, Daratumumab und Dexamethason (IberDd) mit Daratumumab, Bortezomib und Dexamethason (DVd) bei Teilnehmern mit rezidiviertem oder refraktärem Multiplem Myelom (RRMM).
Organisatorische Daten:
| Prüfplancode: | CC-220-MM-002 |
| ISRCTN: | |
| EudraCT: | 2020-000431-49 |
| Clinicaltrials.gov: | NCT04975997 |
| DRKS: | |
| Sponsor: | Celgene |
| Studienphase: | Phase 3 |
| Status: | Rekrutierung läuft, geplant bis Juni 2026 |
Ziele:
Dies ist eine multizentrische, randomisierte, kontrollierte, offene Phase-3-Studie zum Vergleich der Wirksamkeit und Sicherheit von Iberdomid in Kombination mit Dexamethason und Daratumumab (IberDd) gegenüber Daratumumab, Bortezomib und Dexamethason (DVd) bei Teilnehmern mit rezidiviertem oder refraktärem Multiplem Myelom (RRMM). Die Studie untersucht, ob IberDd das progressionsfreie Überleben bei Patienten mit RRMM signifikant verbessert.
Hintergrund:
Iberdomid ist eine Cereblon-bindende Substanz. Präklinische Studien mit Iberdomid haben gezeigt, dass es stärker an das Protein Cereblon bindet als andere Cereblon-bindende Verbindungen. Erste klinische Studien zeigten vielversprechende Ergebnisse für IberDd bei intensiv vorbehandelten Patienten mit RRMM.
(Quelle: https://www.medmedia.at/im-fokus/multiples-myelom/iberdomid-klinisch-vielversprechend/) (Michael Amatangelo et al, Blood 2018. 132, 1935)
Durchführung:
Die Studie wird in zwei Stadien durchgeführt.
In Stadium 1 werden etwa 200 Teilnehmer nach dem Zufallsprinzip einer der folgenden Behandlungen zugeteilt:
Behandlungsarm A1, A2 und A3 erhält Iberdomid in einer von drei Dosisstufen in Kombination mit Daratumumab und Dexamethason (IberDd).
Behandlungsarm B erhält Daratumumab, Bortezomib und Dexamethason (DVd).
In Stadium 2 werden etwa 664 weitere Teilnehmer im Verhältnis 1:1 Behandlungsarm A (IberDd) oder B (DVd)zugeteilt.
Daratumumab und Bortezomib werden als Injektion unter die Haut (subkutan) verabreicht. Iberdomid und Dexamethason werden eingenommen (oral).
In beiden Behandlungsarmen werden die Teilnehmer so lange behandelt, bis ein Fortschreiten der Krankheit (Krankheitsprogression) bestätigt wird, eine nicht vertretbare Nebenwirkung auftritt oder sie ihre Einwilligung zurückziehen. Um die Genauigkeit und Vollständigkeit der Bewertung des progressionsfreien Überlebens zu gewährleisten, werden Teilnehmer, die die Studienbehandlung aus einem anderen Grund als einer bestätigten Krankheit oder der Rücknahme der Einwilligung dauerhaft abbrechen, zur Bewertung der Krankheit weiter beobachtet.
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- Dokumentierte Diagnose eines Multiplen Myeloms (MM) und messbare Krankheit, definiert als
- M-Protein in Serum-Elektrophorese ≥ 1 g/dl und/oder Urinprotein-Elektrophorese ≥ 200 mg/24 Stunden und/oder
- Leichtketten-Myelom ohne messbare Erkrankung im Serum oder Urin.
- Mindestens 1 bis 2 vorherige Myelombehandlungen (Hinweis: Die Induktion mit oder ohne Knochenmarktransplantation und mit oder ohne Erhaltungstherapie gilt als eine Behandlung).
- Ansprechen auf mindestens eine vorherige Behandlung des Myeloms (partielles Ansprechen [PR] oder besser).
Ausschlusskriterien:
- Bestimmte Laborwerte, die nicht im Normalbereich liegen
- Plasmazellleukämie, Waldenström-Makroglobulinämie oder POEMS-Syndrom (Polyneuropathie, Organomegalie, Endokrinopathie, monoklonales Protein und Hautveränderungen) oder klinisch signifikante Amyloidose.
- Periphere Neuropathie Grad 3, Grad 4 oder Grad 2 mit Schmerzen.
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt
Gemeinschaftspraxis Dr. med. Karl Verpoort, Dr. med. Jan Wierecky, PD Dr. med. Wolfgang Zeller
040 - 35 71 777 0
Zusätzliche Information
Fachärzte für Innere Medizin, Hämatologie, internistische Onkologie und Palliativmedizin
Hämato-Onkologische Praxis Dr. med A. Bakhshandeh-Bath
(040) 27 88 91 - 40
Zusätzliche Information
HOPA-Hämatologisch-Onkologische Praxis Altona MVZ GmbH
040-38 02 12 – 60
Katholisches Marienkrankenhaus Gmbh, Zentrum für Innere Medizin
040/25 46-25 02
Universitätsklinikum Eppendorf
040-7410-0
Studien
ABBV-838Dosis-Eskalationsstudie mit ABBV-838, einem Antikörperkonjugat, bei Patienten mit rezidiviertem oder refraktärem multipen Myelom
| Prüfplancode: | M-14-467 |
| ISRCTN: | |
| EudraCT: | 2014-002609-39 |
| Clincaltrials.gov: | NCT02462525 |
| DRKS: |
Allgemeine Informationen
Es wird eine experimentelle Substanz in der Phase I/1b in ansteigenden Dosierungen auf ihre Wirksamkeit und Sicherheit als Einzelsubstanz bei schwer vorbehandelten Myelompatienten geprüft.
Offene Phase-I/II-Multikohortenstudie zur Bewertung der Wirksamkeit und Sicherheit von Cevostamab bei Patienten mit rezidiviertem/refraktärem Multiplem Myelom, die zuvor ein Anti-B-Zell-Reifungsantigen erhalten haben
Organisatorische Daten:
| Prüfplancode: | CO43476 |
| ISRCTN: | |
| EudraCT: | 2021-006816-10 |
| Clinicaltrials.gov: | NCT05535224 |
| DRKS: | |
| Sponsor: | Hoffmann La Roche |
| Studienphase: | Phase 1 |
| Status: | Rekrutierung geplant bis Febr. 2027 |
Hintergrund:
Cevostamab ist ein neuer, gegen zwei Ziele gerichteter (bispezifischer) Antikörper. Er zielt auf einen Rezeptor, der ausschließlich auf Zellen der B-Linie exprimiert wird. Bei stark vorbehandelten Patienten mit rezidiviertem/refraktärem Multiplem Myelom (RRMM) hat Cevostamab eine vielversprechende Wirkung gezeigt. In dieser Studie soll die Wirksamkeit und Sicherheit von Cevostamab als mögliche Therapie für Multiples Myelom weiter untersucht werden. Die Studie wird in unterschiedlichen Kohorten durchgeführt.
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- Dokumentierte Diagnose eines Multiplen Myeloms auf der Grundlage der Standardkriterien der International Myeloma Working Group (IMWG)
- Nachweis eines Fortschreitens der Erkrankung auf der Grundlage der Bestimmung des Ansprechens durch die Prüfer nach den IMWG-Kriterien bei oder nach der letzten Dosierung
- Vorangegangene BCMA-ADC- oder CAR-T-Kohorte: Teilnehmer, die eine gegen BCMA gerichtete Therapie mit Chimären Antigenrezeptor-T-Zellen (CAR-T-Zellen) oder Antikörper-Wirkstoff-Konjugat (ADC)) erhalten haben und auf eine Dreifachtherapie nicht oder nicht mehr ansprechen (refraktär sind)
- Vorangegangene BCMA Bispezifische Kohorte: Teilnehmer, die einen gegen BCMA gerichteten T-Zell-abhängigen bispezifischen (TDB) Antikörper erhalten haben und auf eine Dreifachtherapie nicht oder nicht mehr ansprechen (refraktär sind)
- Messbare Krankheit gemäß den Diagnosekriterien der International Myeloma Working Group (IMWG)
- ECOG- Leistungsstatus (Index zur Abstufung der Lebensqualität von Patienten mit Krebserkrankungen) von 0 oder 1
Ausschlußkriterien:
- Unfähigkeit, den nach dem Protokoll vorgeschriebenen Krankenhausaufenthalt einzuhalten
- Schwangerschaft oder Stillen oder die Absicht, während der Studie oder innerhalb von 5 Monaten nach der letzten Dosis von Cevostamab oder Tocilizumab oder innerhalb von 3 Monaten nach der letzten Dosis von Tocilizumab schwanger zu werden (falls zutreffend)
- Vorherige Behandlung mit Cevostamab oder einem anderen Wirkstoff, der gegen dasselbe Ziel gerichtet ist
- Vorherige BCMA-ADC- oder CAR-T-Kohorte: vorherige Behandlung mit einem beliebigen T-Zell-abhängigen bispezifischen Antikörper (TDB-Antikörper), einschließlich TDB, die nicht auf BCMA abzielen
- Vorherige Anwendung eines monoklonalen Antikörpers, eines Radioimmunkonjugats oder eines ADC als Krebstherapie innerhalb von 4 Wochen vor der ersten Studienbehandlung, ausgenommen die Anwendung einer Nicht-Myelom-Therapie
- Vorherige Behandlung mit systemischen Immuntherapeutika
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Studie mit Iberdomid
Eine Studie zur Bestimmung der Dosis und Verträglichkeit der CC-220 Monotherapie mit Iberdomid, in Kombination mit Dexamethason und in Kombination mit Dexamethason und Daratumumab oder Bortezomib bei Patienten mit fortgeschrittenem und refraktärem Multiplem Myelom (RRMM)
Organisatorische Daten:
| Prüfplancode: | CC-220-MM-001 |
| ISRCTN: | |
| EudraCT: | 2016-000860-40 |
| Clinicaltrials.gov: | NCT02773030 |
| DRKS: | |
| Sponsor: | Celgene |
| Studienphase: | Phase 1b/2a |
| Status: | Rekrutierung läuft, geplant bis Mai 2026 |
Ziel:
Dies ist eine multizentrische, länderübergreifende, offene Phase-1b/2a-Dosiseskalationsstudie zur Bestimmung der höchsten verträglichen Dosis, der Sicherheit, Verträglichkeit, Pharmakokinetik (Einwirkung des Körpers auf die Substanz)und Wirksamkeit von CC-220 (Iberdomid) als Monotherapie und in Kombination mit anderen Therapien bei Patienten mit rezidivierendem oder therapierefraktärem Multiplem Myelom.
Hintergrund:
Iberdomid (CC-220) ist eine neuartige Substanz, die in dieser Phase I/II Studie zur Behandlung von fortgeschrittenem und refraktärem Multiplem Myelom in Kombination mit anderen Therapien untersucht wird. Präklinische Studien zu Iberdomid zeigen, dass es wirksamer an das Protein Cereblon bindet als andere Cereblon-bindende Substanzen. Klinische Studien zu Bortezomib und Daratumumab in Kombination mit anderen Cereblon-bindenden Mitteln haben eine hohe Verträglichkeit bei bemerkenswerter Wirksamkeit bei RRMM gezeigt. Allerdings wurden diese Kombinationen mit Iberdomid bisher nicht untersucht. Insgesamt gesehen untermauern die vorliegenden präklinischen Daten die Untersuchung von Iberdomid in Kombination mit sowohl Bortezomib/Dexamethason als auch Daratumumab in klinischen Studien.
(Michael Amatangelo et al, Blood 2018. 132, 1935; http://www.bloodjournal.org/content/132/Suppl_1/1935?sso-checked=true
Durchführung:
Die Studie wird in zwei Teilen durchgeführt: Teil 1 mit Kohorte A und B und Teil 2 mit Kohorte C, D, E, F, G1 und G2.
In Teil 1 (Phase 1b) wird die maximal verträgliche Dosis von oralem CC-220 als Monotherapie (Kohorte A) und die von oralem CC-220 in Kombination mit oralem Dexamethason (Kohorte B) bestimmt. Außerdem wird in diesem Teil die empfohlene CC-220-Dosis für Teil 2 (Phase 2) ermittelt.
In Teil 2 werden folgende Therapien untersucht:
Kohorte C: CC-220 als Monotherapie in der empfohlenen Dosis für Teil 2
Kohorte D: CC-220 in der empfohlenen Dosis für Teil 2 in Kombination mit oralem Dexamethason
Kohorte E:CC-220 in Kombination mit oralem Dexamethason und intravenösem Daratumumab
Kohorte F: CC-220 in Kombination mit oralem Dexamethason und subkutanem Bortezomib
Kohorte G1: CC-220 in Kombination mit einmal wöchentlich verabreichtem intravenösem Carfilzomib und oralem Dexamethason
Kohorte G2: CC-220 in Kombination mit zweimal wöchentlich verabreichtem intravenösem Carfilzomib und oralem Dexamethason.
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- Dokumentierte Diagnose eines Multiplen Myeloms (MM) und messbare Krankheit, definiert als
- M-Protein: Serum-Elektrophorese ≥ 0,5 g/dl und/oder Urinprotein-Elektrophorese ≥ 200 mg/24 Stunden und/oder
- Leichtketten-Myelom ohne messbare Erkrankung im Serum oder Urin - Dokumentierte Krankheitsprogression an oder innerhalb von 60 Tagen seit der letzten Dosis der letzten Myelombehandlung
- ECOG (Index zur Abstufung der Lebensqualität von Patienten mit Krebserkrankungen) von 0, 1 oder 2
Ausschlußkriterien:
- Nicht ekretorisches oder oligosekretorisches Multiples Myelom
- Plasmazellenleukämie oder Amyloidose
- Bestimmte Laborwerte, die nicht im Normalbereich liegen
- Periphere Neuropathie ≥ Grad 2
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Ein neuer Cereblon E3 Ligase Modulator
Eine Studie zur Bestimmung der empfohlenen Dosis und des Regimes und zur Bewertung der Sicherheit und vorläufigen Wirksamkeit von CC-92480 in Kombination mit Standardbehandlungen bei Patienten mit rezidiviertem oder refraktärem Multiplen Myelom (RRMM) und neu diagnostiziertem Multiplen Myelom (NDMM)
Organisatorische Daten:
| Prüfplancode: |
CC-92480-mm-02 |
| UTMS | U1111-1233-5619 |
| EudraCT: | 2018-004767-31 |
| Clinicaltrials.gov: | NCT03989414 |
| DRKS: | |
| Sponsor: | Celgene |
| Studienphase: | Phase 1/2 |
| Status: | Rekrutierung läuft, geplant bis Januar 2025 |
Hintergrund:
CC-92480 ist ein sogenannter CRBN-Modulator [Cereblon-(CRBN-)E3-Ligase-Modulator (CELMoD)]. Dieser neuartige Modulator hat vielfältige Wirkungen und wirkt unter anderem stark immunmodulierend. Der Wirkstoff führt zu einem schnellen, tiefen und anhaltenden Zerfall von Ikaros und Aiolos – zwei Faktoren, die zum Überleben der Myelomzellen beitragen.
Durchführung:
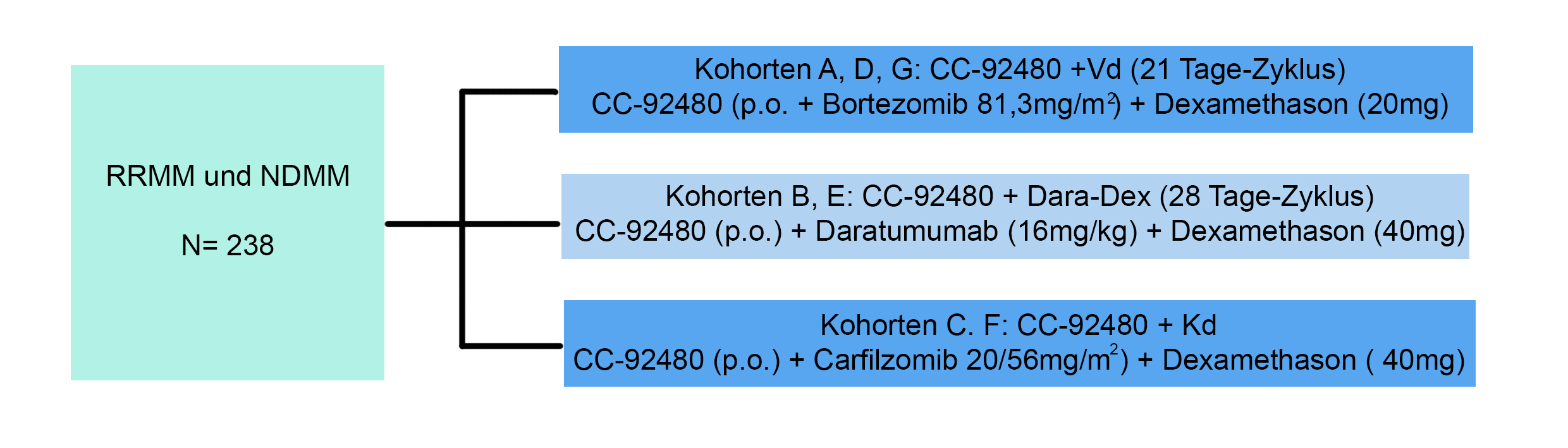
Die Studie wird randomisiert ausgeführt und die Patienten in 3 Gruppen eingeteilt, die neben dem CC-92480 unterschiedliche Zweier-Kombinationen enthalten, die gegeneinander getestet werden. Die Kohorten haben unterschiedliche Einschlusskriterien.
Ein und Ausschlusskriterien:
Einschlusskriterien:
- Alter ≥ 18 Jahre alt und ECOG (Index zur Abstufung der Lebensqualität von Patienten mit Krebserkrankungen) mit 0, 1 oder 2 Punkten.
- Patienten mit rezidiviertem oder refraktäremMultiplen Myelom müssen eine messbare Krankheit haben und ihr Krankheitsverlauf während oder nach ihrer letzten Myelomtherapie muss dokumentiert sein.
- Bei neu diagnostizierten Patienten muss die Diagnose eines zuvor unbehandelten symptomatischen Multiplen Myeloms dokumentiert sein.
- Frauen im gebärfähigen Alter und männliche Patienten müssen mit dem Schwangerschaftsverhütungsplan einverstanden sein.
Ausschlusskriterien:
- Signifikante medizinische Erkrankung, auffällige Laborwerte oder eine psychiatrische Krankheit, die an der Teilnahme an der Studie hindert.
- Patient ist nicht in der Lage oder nicht bereit, sich der laut Prüfplan vorgeschriebenen Prophylaxe einer Thromboembolie zu unterziehen.
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt
Studie zu CC-93269, einem BCMA x CD3 T-Zell-aktivierendem Antikörper, bei Patienten mit rezidivierendem und refraktärem multiplem Myelom
Organisatorische Daten:
| Prüfplancode: |
CC-93269 |
| ISRCTN: | |
| EudraCT: | |
| Clinicaltrials.gov: | NCT03486067 |
| DRKS: | |
| Sponsor: | Celgene |
| Studienphase: | Phase 1 |
| Status: | Rekrutierung läuft, geplant bis Juli 2027 |
Kurzbeschreibung:
In der offenen Phase-1-Studie CC-93269-MM-001 wird CC-93269 erstmals beim Menschen – und zwar bei Patienten mit rezidiviertem und refraktärem multiplem Myelom – untersucht.
Hintergrund:
CC-93269 ist ein sogenannter bispezifischer T-Zell Antikörper (BiTE Antikörper= Bi-specific T-cell engagers). Diese Antikörper verknüpfen das auf Myelomzellen vorhandene B-Zell-Reifungsantigen (BCMA) mit T-Zellen und führen so zu einer T-Zell vermittelten Zerstörung von Myelomzellen. BCMA, ein Mitglied der Tumornekrosefaktor-Rezeptor-Superfamilie, das auf malignen Plasmazellen vermehrt vertreten ist, spielt eine Schlüsselrolle bei der Förderung des Überlebens von Plasmazellen.
Durchführung:
Die Studie besteht aus zwei Teilen: einem Dosissteigerungsteil (Teil A) und einem Erweiterungsteil (Teil B). In Teil A wird die Sicherheit und Verträglichkeit von intravenös verabreichtem CC-93269 in steigenden Dosen bewertet, um die höchste verträgliche Dosis und die nicht verträgliche Dosis sowohl der ersten als auch der nachfolgenden Gaben von CC-93269 zu bestimmen. In Teil B wird die Sicherheit und Wirksamkeit von CC-93269, das den Teilnehmern in ausgewählten Studiengruppen in der höchsten verträglichen oder einer niedrigeren Dosis verabreicht wird, weiter untersucht, um die empfohlene Dosis für die Phase 2 zu bestimmen. Alle Behandlungen werden in 28-Tage-Zyklen bis zu 2 Jahre lang verabreicht, bis die Erkrankung fortschreitet, nicht vertretbare Nebenwirkungen auftreten oder der Teilnehmer/Prüfer sich zum Abbruch der Teilnahme entscheiden.
Einschlusskriterien:
- Multiples Myelom (MM) in der Vorgeschichte mit rezidivierter und refraktärer Erkrankung und erfolglose Behandlung mit, Unverträglichkeit gegenüber oder fehlende Eignung für verfügbare(n) Therapien
-
Messbare Krankheit gemäß Beurteilung durch ein Zentrallabor
-
Einwilligung zum Krankenhausaufenthalt zur Überwachung und zur Entnahme von peripheren Blutproben
-
Einwilligung zu Knochenmarkpunktionen und/oder Gewebeproben im Rahmen der Studie
Ausschlusskriterien:
- Vorbehandlung mit einer in der Erforschung befindlichen Therapie gegen das B-Zell-Reifungsantigen (BCMA)
- Multiples Myelom mit symptomatischer Beteiligung des zentralen Nervensystems
- Nicht-sezernierendes multiples Myelom, Plasmazellleukämie, Morbus Waldenström (Makroglobulinämie), POEMS-Syndrom (Polyneuropathie, Organomegalie, Endokrinopathie, monoklonales Protein und Hautveränderungen) oder Amyloidose
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt
Plattform- Studie mit Belantamab-Mafodotin zum multiplen Myelom
Plattformstudie zu Belantamab-Mafodotin als Monotherapie und in Kombination mit Anti-Krebs-Therapien bei Teilnehmern mit rezidiviertem/refraktärem Multiplem Myelom (RRMM) (DREAMM 5)
Organisatorische Daten:
| Prüfplancode: | 208887 - Dreamm 5 |
| ISRCTN: | |
| EudraCT: | 2019-001138-32 |
| Clinicaltrials.gov: | NCT04126200 |
| DRKS: | |
| Sponsor: | GlaxoSmithKline |
| Studienphase: | 3 |
| Status: | Rekrutierung läuft, geplant bis Februar 2028 |
Kurzbeschreibung:
Bei der DREAMM 5-Studie handelt sich um eine randomisierte, offene Phase-1/2-Plattform-Studie, die die Wirkung von Belantamab-Mafodotin in Kombination mit anderen Krebsmedikamenten bei Teilnehmern mit rezidiviertem/refraktärem multiplem Myelom untersuchen soll. Das Plattform-Design dient dazu, mehrere Behandlungskombinationen gleichzeitig als Sub-Studien im Rahmen eines übergeordneten Prüfplans zu untersuchen.
Ziel:
Die DREAMM 5- Studie zielt darauf ab, in der Dosisfindungsphase eine sichere und verträgliche Dosis für die Erweiterungsphase der Studie zu ermitteln, und dann in dieser Phase die Gesamtansprechrate der Kombinationsbehandlung bzw. Monotherapie mit Belantamab-Mafodotin (bis zu einem Zeitraum von 36 Monaten) zu bestimmen.
Hintergrund:
Die amerikanische (FDA) und europäische (EMA) Arzneimittelbehörde haben Belantamab-Mafodotin-blmf (GSK2857916; Blenrep), basierend auf der DREAMM-2 Studie, für die Behandlung von Patienten mit rezidiviertem oder refraktärem Multiplen Myelom zugelassen, die zuvor mindestens vier Therapien erhalten haben und deren Erkrankung auf die Behandlung mit mindestens einem Proteasom-Inhibitor, einem Immunmodulator und einem monoklonalen Anti-CD38-Antikörper (Arten von Krebsarzneimitteln) nicht anspricht und deren Krebserkrankung sich seit der letzten Therapie verschlechtert hat. Es wird einmal alle drei Wochen als Tropfinfusion in eine Vene gegeben, wobei die Dosis vom Körpergewicht abhängt. Sehr häufige Nebenwirkungen von Blenrep sind Keratopathie (Hornhautschädigung, die mehr als 7 von 10 Behandelten betreffen kann) und Thrombozytopenie (niedrige Anzahl an Blutplättchen, die mehr als 3 von 10 Behandelten betreffen kann). Sehr häufige schwere Nebenwirkungen (die bis zu 1 von 10 Behandelten betreffen können) sind Pneumonie (Lungenentzündung), Fieber und infusionsbedingte Reaktionen. Dies ist weltweit die erste Anti-BCMA-Behandlung, die bei dieser Patientenpopulation verfügbar ist.Der Wirkstoff in Blenrep, Belantamab-Mafodotin, besteht aus einem monoklonalen Antikörper (einer Art von Protein), der an ein zytotoxisches (zelltötendes) Molekül gebunden ist. Der Antikörper wurde so entwickelt, dass er an ein Protein mit der Bezeichnung B-Zell-Reifungsantigen (BCMA) bindet, das auf der Oberfläche anomaler unreifer Plasmazellen (Myelomzellen) vorhanden ist. Wenn der Patient Blenrep erhält, bindet der Antikörper-Anteil des Arzneimittels an das BCMA auf den Myelomzellen und gibt das zytotoxische Molekül in die Zellen ab. Im Zellinneren tötet das zytotoxische Molekül die Zellen ab, indem es ihr Teilungs- und Wachstumsvermögen hemmt. Blenrep regt außerdem das Immunsystem (die natürliche Abwehr des Körpers) dazu an, die Myelomzellen anzugreifen. In der Kombination sollen diese Wirkungen das Fortschreiten der Erkrankung verlangsamen.
Einschlusskriterien:
- ECOG (Index zur Abstufung der Lebensqualität von Patienten mit Krebserkrankungen) von 0 bis 1, es sei denn, ein ECOG-Wert von bis zu 2 ist ausschließlich auf MM-bedingte Skelettprobleme und/oder Skelettschmerzen zurückzuführen.
- Histologisch oder zytologisch bestätigte Diagnose eines Multiplen Myeloms (MM) gemäß der Definition der IMWG.
- Vorbehandlung des Multiplen Myeloms mit mindestens 3 Therapielinien, einschließlich eines immunmodulierenden Wirkstoffs (IMID), eines Proteasominhibitors (PI) und eines monoklonalen Anti-CD38-Antikörpers.
- Patienten mit einer autologen Stammzelltransplantation in der Vorgeschichte sind zur Studienteilnahme berechtigt, wenn die Transplantation > 100 Tage vor Studieneinschluss erfolgte und keine aktive(n) Infektion(en) vorliegen.
Ausschlusskriterien
- Aktuelle Erkrankung des Hornhautepithels außer leichter punktueller Keratopathie (Hornhauterkrankung).
- Anzeichen eines kardiovaskulären Risikos
- Bekannte unmittelbare oder verzögerte Überempfindlichkeitsreaktion auf Medikamente, die chemisch mit Belantamab-Mafodotin oder einem der Bestandteile der Studienmedikation verwandt sind. Vorgeschichte einer schweren Überempfindlichkeit gegen andere monoklonale Antikörper.
- Aktive Infektion, die eine antibiotische, antivirale oder antimykotische Behandlung erfordert.
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Belantamab-Mafodotin in Kombination mit Bortezomib + Lenalidomid + Dexamethason
Organisatorische Daten:
| Prüfplancode: | 209664 |
| ISRCTN: | |
| EudraCT: | 2019-003047-30 |
| Clinicaltrials.gov: | NCT04091126 |
| DRKS: | |
| Sponsor: | GlaxoSmithKline |
| Studienphase: | I |
| Status: | Rekrutierung geplant bis 11/2025 |
Ziel:
In dieser randomisierten, offenen Phase-I-Studie werden die Wirksamkeit und Sicherheit von Belantamab-Mafodotin in Kombination mit Bortezomib, Lenalidomid und Dexamethason (VRd) im Vergleich zu VRd allein bei Patienten mit neu neudiagnostiziertem Multiplen Myelom untersucht, die nicht für eine autologe Stammzelltransplantation geeignet sind.
Durchführung:
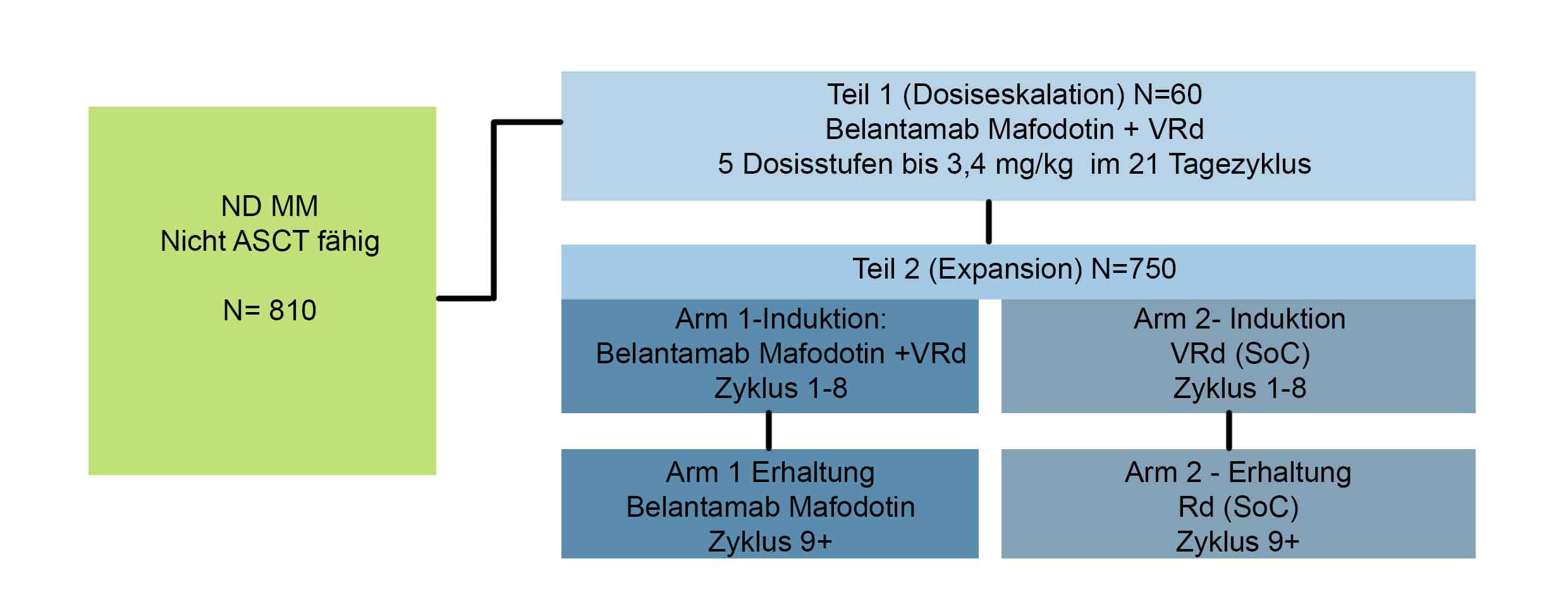
Die Studie besteht aus zwei Teilen:
In Teil 1 werden die Sicherheit und Verträglichkeit von Belantamab-Mafodotin in bis zu 5 Dosisstufen in Kombination mit VRd bewertet und die empfohlene Dosis für die Phase III (Teil 2) ermittelt.
In Teil 2 (Verlängerung) werden die Wirksamkeit und Sicherheit der für die Phase III empfohlenen Dosis von Belantamab-Mafodotin in Kombination mit VRd im Vergleich zu VRd allein bewertet.
Hintergrund:
Belantamab-Mafodotin (vormals GSK2857916) ist ein gegen das Oberflächenantigen BCMA (engl. B-Cell Maturation Antigen) gerichtetes Antikörper-Wirkstoff-Konjugat. Sobald Belantamab an BCMA bindet, wird das krebstötende Molekül in die Zelle freigesetzt und führt den Zelltod herbei. Bei stark vortherapierten Patienten mit rezidiviertem/refraktärem multiplem Myelom hat Belantamab-Mafodotin eine hohe Wirksamkeit gezeigt. Weitere Untersuchungen müssen zeigen, inwieweit auftretende Nebenwirkungen, teilweise mit vorübergehender Beeinträchtigung des Sehvermögens, die Einsatzmöglichkeiten dieser Therapie einschränken. Vielversprechend erscheinen Kombinationen mit etablierten Substanzen zur weiteren Verbesserung der Ansprechrate, -tiefe, und -dauer.
Belantamab-Mafodotin ist noch nicht zur Anwendung zugelassen. Allerdings wird der Antrag auf Zulassung des Arzneimittels für die Behandlung von Patienten mit rezidiviertem oder therapierefraktärem Multiplen Myelom im Rahmen eines beschleunigten Verfahrens derzeit sowohl von der amerikanischen Zulassungsbehörde FDA als auch der europäischen Arzneimittelbehörde EMA geprüft. (Stand der Information: Februar 2020, https://myelomaresearchnews.com/2020/02/05/ema-review-gsks-belantamab-mafodotin-treatment-heavily-pretreated-multiple-myeloma/
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
-
ECOG Performance Status (Index zur Abstufung der Lebensqualität von Patienten mit Krebserkrankungen) von 0 bis2
-
Neudiagnose Multiples Myelom
-
Der Patient ist für die Hochdosistherapie und die autologe Stammzelltransplantation nicht geeignet
-
Der Patient erfüllt mindestens eines der folgenden Kriterien einer messbaren Krankheitsaktivität:
Urin-Leichtkette (M-Protein) ≥ 200 mg/24 Stunden ≥ 0,2 g/24 Stunden) oder
Serum M-Protein ≥ 0,5 g/l ≥ 5,0 g/l]) oder
Konzentration der betroffenen freien Leichtkette im Serum (sFLC) ≥ 10 mg/dl (≥ 100 mg/l) und abnormes sFLC-Verhältnis (< 0,26 oder > 1,65).
Ausschlußkriterien:
- Smouldering Myelom (SMM)
- Systemische Vortherapie (mit Ausnahme von insgesamt bis zu 160 mg Dexamethason über 4 Tage)
- Periphere Neuropathie ≥ Grad
- Größere Operation innerhalb von 4 Wochen vor der ersten Gabe der Studienmedikation
- Keratopathie
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Induktionstherapie mit Lenalidomid, Bortezomib und Dexamethason und entweder intravenösem oder subkutanem Isatuximab bei Patienten mit neu diagnostiziertem Multiplem Myelom, die für eine Hochdosis-Chemotherapie mit nachfolgender autologer Stammzelltransplantation geeignet sind
Organisatorische Daten:
| Prüfplancode: | GMMG-HD 8 / DSMM XIX |
| ISRCTN: | |
| EudraCT: | 2022-000996-38 |
| Clinicaltrials.gov: | NCT05804032 |
| DRKS: | |
| Sponsor: | Universitätsklinikum Heidelberg DSMM, Sanofi und KKS-Netzwerk |
| Studienphase: | |
| Status: | Rekrutierung geplant bis Juni 2025 |
Ziel:
In dieser Studie für Patienten mit zuvor unbehandeltem Multiplem Myelom soll die Wirksamkeit der Induktionstherapie mit Isatuximab in Kombination mit Lenalidomid/Bortezomib/Dexamethason (RVd) bei Gabe von Isatuximab unter die Haut (subkutan) gegenüber einer Gabe in die Vene (intravenös) untersucht werden.
Durchführung:
Randomisierung: Die Patienten werden vor der Induktionstherapie nach dem Zufallsverfahren (randomisiert) einem von 2 Studienarmen (A oder B) zugeordnet.
- Patienten in Arm A erhalten 3 Zyklen des monoklonalen Antikörpers Isatuximab intravenös in Kombination mit dem RVd-Schema (Lenalidomid, Bortezomib, Dexamethason). Jeder Zyklus hat eine Dauer von 42 Tagen.
- Patienten in Arm B erhalten 3 Zyklen Isatuximab subkutan in Kombination mit RVd.
Nach der Induktionstherapie erhalten die Patienten eine Standard-Intensivierung (in der Regel eine Mobilisierungstherapie auf Cyclophosphamid-Basis, Stammzellenentnahme und hochdosiertes Melphalan, gefolgt von einer autologen Stammzelltransplantation
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- Bestätigte Diagnose eines neu diagnostizierten behandlungsbedürftigen Multiplen Myeloms gemäß den überarbeiteten Diagnosekriterien der International Myeloma Working Group (IMWG))
- Der Patient ist für eine Hochdosis-Chemotherapie und eine autologe Stammzelltransplantation geeignet
- Messbare Krankheitsaktivität (gemäß der Definition im Prüfplan)
- Alter 18 bis 70 Jahre bei Studieneinschluss
Ausschlußkriterien:
- Bekannte Überempfindlichkeit (oder Kontraindikation) gegen einen der Bestandteile der Studientherapie
- Systemische Amyloid-Leichtketten-Amyloidose (außer lokalisierte Amyloid-Leichtketten -Amyloidose, die auf die Haut oder das Knochenmark beschränkt ist)
- Plasmazell-Leukämie
- Vorherige Chemo- oder Strahlentherapie in den letzten 5 Jahren (außer lokaler Strahlentherapie bei lokalem Fortschreiten des Myeloms)
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
CAR T-Zelltherapie mit bb2121
Eine Phase-2-Studie zur Bestimmung der Wirksamkeit und Sicherheit von bb2121 bei Patienten mit rezidiviertem (wieder aufgetretenem) und refraktärem (nicht auf Behandlung ansprechendem) Multiplem Myelom (RRMM) und bei Patienten mit risikoreichem Multiplem Myelom, bei denen die Krankheit innerhalb von 18 Monaten nach der Erstbehandlung fortgeschritten ist.
Organisatorische Daten:
| Prüfplancode: | KarMMa2 |
| ISRCTN: | |
| EudraCT: | 2018-000264-28 |
| Clinicaltrials.gov: | NCT03601078 |
| DRKS: | |
| Sponsor: | Celgene |
| Studienphase: | Phase 2 |
| Status: | Rekrutierung läuft, geplant bis März 2025 |
Ziel:
In dieser Studie soll die Wirksamkeit und Sicherheit von bb2121 bei Patienten mit rezidiviertem oder refraktären Multiplen Myelom bewertet werden.
Hintergrund:
Die chimäre-Antigenrezeptor-(CAR) T-Zelltherapie bb2121 ist gegen das Oberflächenantigen BCMA (engl. B-cell maturation antigen) gerichtet, um T-Zellen dahin gehend zu programmieren, dass sie maligne Myelomzellen erkennen und abtöten. https://meetinglibrary.asco.org/record/160693/abstract
In einer Phase-1-Studie bei stark vorbehandelten Patienten mit rezidiviertem oder refraktären multiplem Myelom zeigte bb2121 ermutigende Ergebnisse mit tiefem und dauerhaftem Ansprechen bei gleichzeitig guter Verträglichkeit. https://www.presseportal.de/pm/62626/3972298 In der vorliegenden Studie soll die Wirksamkeit und Sicherheit von bb2121 als mögliche Therapie für Multiples Myelom weiter untersucht werden.
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- Es besteht eine messbare Krankheit, definiert als
- M-Protein (Serum-Protein-Elektrophorese [sPEP] oder Urin-Protein-Elektrophorese [uPEP]): sPEP ≥ 0,5 g/dl oder uPEP ≥ 200 mg/24 Stunden und/oder
- Leichtketten-MM ohne messbare Krankheit im Serum oder Urin: freie Leichtketten im Serum ≥ 10 mg/dl und abnormes Kappa-Lambda-Leichtketten-Verhältnis im Serum
- Spezifische Anforderungen für:
- Kohorte 1: ≥ 3 frühere Anti-Myelom-Behandlungsschemen
- Kohorte 2: 1 frühere Anti-Myelom-Behandlung
- ECOG Performance Status von 0 oder 1 ((Index zur Abstufung der Lebensqualität von Patienten mit Krebserkrankungen)
Ausschlusskriterien:
- Erhalt eines Prüfpräparats innerhalb von 14 Tagen vor der Leukapherese
- Eines der folgenden Verfahren innerhalb der letzten 14 Tage vor der Leukapherese:
- Plasmapherese
- Große Operation (nach Definition des Prüfarztes)
- Strahlentherapie mit Ausnahme der lokalen Therapie für myelombedingten Knochenläsionen
- Anwendung einer systemischen Anti-Myelom-Medikamententherapie
- Bekannte Beteiligung des zentralen Nervensystems
- Klinischer Nachweis einer Lungenleukostase und Verbrauchskoagulopathie (disseminierten intravasale Gerinnung)
- Vorgeschichte oder Vorliegen einer klinisch relevanten Erkrankung des Zentralnervensystems (ZNS)
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Ergänzende Informationen
Zur Herstellung der mit einem bb2121 chimären Antigenrezeptor (CAR) modifizierten T-Zellen werden die weißen Blutkörperchen (Leukozyten) aus dem Blut herausgelöst. Dies bezeichnet man als Leukapherese. Vor der bb2121-Infusion wird bei den Patienten ein Verfahren zur Entfernung der Lymphozyten, die sogenannte Lymphozytendepletion, mit Fludarabin und Cyclophosphamid durchgeführt.
Intravenös verabreichtes ABBV-383 in Kombination mit Krebstherapien bei Patienten mit rezidiviertem oder refraktärem Multiplen Myelom: eine Studie zur Bewertung der unerwünschten Ereignisse und der Veränderung der Krankheitsaktivität
Organisatorische Daten:
NCT05259839
| Prüfplancode: |
M22-947 |
| ISRCTN: | |
| EudraCT: | |
| Clinicaltrials.gov: | NCT05259839 |
| DRKS: | |
| Sponsor: | TeneoOne Inc |
| Studienphase: | Phase 1, Erstanwendung am Menschen |
| Status: | Rekrutierung läuft, geplant bis Okt.2028 |
Ziele:
Das Multiple Myelom (MM) ist eine Plasmazellerkrankung, die durch das Wachstum klonaler Plasmazellen im Knochenmark gekennzeichnet ist. Ziel dieser Studie ist es, die Sicherheit und Toxizität von ABBV-383 bei gleichzeitiger Verabreichung mit Pomalidomid-Dexamethason (Pd), Lenalidomid-Dexamethason (Rd), Daratumumab-Dexamethason (Dd) oder Nirogacestat (Niro) bei erwachsenen Teilnehmern mit rezidiviertem/refraktärem Multiplem Myelom (MM) zu untersuchen. Es werden unerwünschte Ereignisse und Veränderungen der Krankheitsaktivität bewertet.
Durchführung:
In dieser Studie werden die Teilnehmer in Gruppen eingeteilt, die als Behandlungsarme bezeichnet werden. Untersucht wird ABBV-383 in Kombination mit Pd, Rd, Dd oder Niro. Jeder Behandlungsarm erhält eine andere Behandlungskombination, je nach Stadium der Studie und Eignung des Teilnehmers. Die Studie umfasst eine Dosissteigerungsphase, in der die beste Dosis von ABB-383 ermittelt werden soll. Anschließend folgt eine Dosiserweiterungsphase, in der die Dosis bestätigt werden soll.. Ungefähr 270 erwachsene Teilnehmer mit rezidiviertem/refraktärem MM werden an etwa 45 Standorten weltweit in die Studie aufgenommen. Die Teilnehmer erhalten ABBV-383 intravenös (i.v.) in Kombination mit oral/i.v. verabreichtem Pd, oral/i.v. verabreichtem Rd, oral/i.v./subkutan (s.c.) verabreichtem Dd oder oral/i.v. verabreichtem Niro in 21-tägigen Zyklen.
Für die Teilnehmer an dieser Studie kann die Belastung durch die Behandlung höher sein als bei ihrer Standardbehandlung. Während der Studie werden die Teilnehmer regelmäßig untersucht und die Wirkung und etwaige Nebenwirkungen der Behandlung werden häufig durch medizinische Beurteilungen und Blutuntersuchungen und Fragebögen überprüft.
Hintergrund:
ABBV-383 ist ein Prüfpräparat, das für die Behandlung des rezidivierenden/refraktären Multiplen Myeloms entwickelt wird. Es ist ein Antikörper, der (wie Elranatamab und Teclistamab) gegen zwei Ziele gerichtet (bispezifisch) ist: gegen BCMA und gegen CD3. BCMA ist ein Zelloberflächenprotein, das besonders auf Myelom- und Plasmazellen vorkommt und eine besondere biologische Bedeutung für das Überleben der Plasmazelle hat. Es wird bei Menschen mit Multiplem Myelom in signifikant höherem Maße exprimiert. CD3 ist an der Aktivierung der Immunantwort zur Bekämpfung von Infektionen beteiligt.
.
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- Dokumentierte Diagnose eines Multiplen Myeloms gemäß den IMWG-Diagnosekriterien
- ECOG (Index zur Abstufung der Lebensqualität von Patienten mit Krebserkrankungen) von 0, 1 oder2
- Es muss eine bestätigte Diagnose eines rezidivierten/refraktären Multiplen Myeloms (MM) mit dokumentierten Hinweisen auf ein Fortschreiten der Erkrankung während oder nach der letzten Behandlung des Teilnehmers vorliegen, wie vom Prüfarzt anhand der Kriterien der International Myeloma Working Group (IMWG) ermittelt wurde.
- Es muss eine messbare Erkrankung vorliegen, wie im Prüfplan dargelegt.
- Teilnehmer sind bislang nicht mit ABBV-383 vorbehandelt und haben noch nie eine BCMA-gerichtete Therapie erhalten. Teilnehmer, die eine zielgerichtete Therapie gegen Nicht-BCMA-Targets erhalten haben, werden nicht ausgeschlossen.
Ausschlusskriterien:
- Periphere autologe Stammzelltransplantation (SZT) innerhalb von 12 Wochen oder allogene SZT innerhalb eines Jahres vor der ersten Gabe des Studienmedikaments.
- Fortbestehende unerwünschte Ereignisse ≥ Grad 2 (National Cancer Institute [NCI] Common Terminology Criteria for Adverse Events [CTCAE] Version 5.0) infolge einer früheren Krebstherapie.
- Bekannter Myelombefall des zentralen Nervensystems.
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt
Phase 3- Studie zu Teclistamab
Eine Studie zu Teclistamab in Kombination mit Lenalidomid und Teclistamab allein im Vergleich zu Lenalidomid allein bei Teilnehmern mit neu diagnostiziertem Multiplem Myelom als Erhaltungstherapie nach autologer Stammzelltransplantation (MajesTEC-4)
Organisatorische Daten:
| Prüfplancode: | EMN30/64007957MMY3003 |
| ISRCTN: | |
| EudraCT: | |
| Clinicaltrials.gov: | NCT05243797 |
| DRKS: | |
| Sponsor: | Janssen |
| Studienphase: | Phase 3 |
| Status: | Rekrutierung läuft, geplant bis Oktober 2028 |
Ziele:
Ziel dieser Studie ist es, den Nutzen von Teclistamab in Kombination mit Lenalidomid und von Teclistamab als Monotherapie im Vergleich zu einer Monotherapie mit Lenalidomid bei Patienten mit Multiplem Myelom nach einer autologen Stammzelltherapie zu überprüfen.
Hintergrund:
Teclistamab ist ein bispezifischer Antikörper, der sowohl gegen das B-Zell-Reifungsantigen (BCMA), ein Protein auf Myelomzellen, als auch gegen CD3-Rezeptoren auf der T-Zell-Oberfläche gerichtet ist. Teclistamab wurde im August 2022 in der EU als Monotherapie zur Behandlung des fortgeschrittenen Multiplen Myeloms zugelassen und wird jetzt bei weiteren Indikationen geprüft.
In der MajesTEC-1-Studie bei stark vorbehandelten Patienten mit Multiplem Myelom (n=165) bewirkte Teclistamab ein hohes und dauerhaftes Ansprechen. Bei einer medianen Nachbeobachtungszeit von etwa 14 Monaten betrug die Gesamtansprechrate 63 Prozent, wobei 39,4 Prozent ein komplettes Ansprechen oder besser erreichten. Diese Daten wurden auf der Jahrestagung 2022 der American Society of Clinical Oncology (ASCO) veröffentlicht.
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- Neu diagnostiziertes Multiples Myelom gemäß den Diagnosekriterien der International Myeloma Working Group (IMWG) und Erhalt einer Induktionstherapie mit oder ohne Konsolidierung
- Erhalt nur einer Therapielinie und Erreichen eines mindestens partiellen Ansprechens gemäß den IMWG-Ansprechkriterien ohne Anzeichen einer Progression zum Zeitpunkt der ersten Gabe der Studienmedikation
- ECOG-Performance-Status-Score (Index zur Abstufung der Lebensqualität von Patienten mit Krebserkrankungen) von 0, 1 oder 2 beim Screening und unmittelbar vor Beginn der Verabreichung der Studienmedikation
Ausschlusskriterien:
- Unverträglichkeit gegenüber der Anfangsdosis von Lenalidomid
- Erhaltungstherapie
- Vorherige gegen das B-Zell-Reifungsantigen (BCMA) gerichtete Therapie
- Vorherige Therapie mit einem Mittel, das Immunzellen neu ausrichtet, oder einer gentechnisch veränderten adoptiven Zelltherapie (z. B. mit chimärem Antigenrezeptor-modifizierte T-Zellen, NK-Zellen)
- Abbruch der Behandlung aufgrund einer Nebenwirkung von Lenalidomid, bei der der Prüfarzt einen Zusammenhang mit Lenalidomid sieht
- Fortschreiten des Multiplen Myeloms zu irgendeinem Zeitpunkt vor dem Screening
- Erhalt einer kumulativen Kortikosteroid-Dosis, die ≥ 140 mg Prednison entspricht, in den 14 Tagen vor der ersten Behandlungsdosis
- Impfung mit einem abgeschwächten Lebendimpfstoff innerhalb von 4 Wochen vor der ersten Behandlungsdosis. Totimpfstoffe und nicht replizierende, für den Notfall zugelassene Impfstoffe sind erlaubt.
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt
Eine Studie zur Kombination von Selinexor, Pomalidomid und Dexamethason (SPd) gegenüber Elotuzumab, Pomalidomid und Dexamethason (EloPd) bei Patienten mit vorbehandeltem Multiplem Myelom
Organisatorische Daten:
| Prüfplancode: | EMN29 | |
| ISRCTN: | ||
| EudraCT: | ||
| Clinicaltrials.gov: | NCT05028348 | |
| DRKS: | ||
| Sponsor: |
|
|
| Studienphase: | 3 | |
| Status: | Rekrutierung geplant bis Juli 2023 |
Ziel:
Ziel der Studie ist ein Vergleich zwischen Selinexor und Elotuzumab, jeweils in Kombination mit Pomalidomid und Dexamethason, in Hinblick auf die Wirksamkeit, Sicherheit und gesundheitsbezogene Lebensqualität bei Patienten mit rezidiviertem oder refraktärem Multiplem Myelom.
Hintergrund:
Selinexor ist ein als Tablette verabreichter Wirkstoff, der das Protein Exportin 1 (XPO1) hemmt. Dieses ist ein Schlüsselprotein beim Transport von Molekülen aus dem Zellkern. Mithilfe von XPO1 verteidigen sich Krebszellen gegen Tumorsuppressor-Proteine, die vom Körper zur Krebsbekämpfung eingesetzt werden. Die Suppressoren dringen in den Zellkern der Krebszelle ein und unterdrücken dort entweder deren Fortpflanzungsfähigkeit oder fördern den frühen Zelltod. XP01 bindet an die Proteine, exportiert sie aus dem Zellkern der Krebszelle und sichert somit deren Überleben. Selinexor stellt die Fähigkeit zur Selbstkontrolle wieder her, indem es an XPO1 bindet und dadurch verhindert, dass die Krebszellen Tumorsuppressor-Proteine aus dem Zellkern hinausschleusen. (Quelle: https://www.pharmazeutische-zeitung.de/neues-wirkprinzip-beim-multiplen-myelom-123488/)
Selinexor (Nexpovio®, Kayropharm) ist als 20-mg-Filmtabletten seit Oktober 2022 zur Behandlung des Multiplen Myeloms auf dem deutschen Markt.
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- Rezidiviertes oder refraktäres MM mit messbarer Erkrankung gemäß IMWG-Leitlinien, wie im Protokoll definiert.
- Die Patienten haben mindestens 1 und nicht mehr als 4 vorherige Anti-MM-Therapielinien erhalten. Eine Induktionstherapie, gefolgt von einer Stammzelltransplantation und einer Konsolidierungs-/Erhaltungstherapie, wird als eine Therapielinie betrachtet.
- Die Patienten müssen eine vorherige Therapie erhalten haben, die einen monoklonalen Anti-CD3-Antikörper und 2 oder mehr aufeinanderfolgende Zyklen der folgenden Wirkstoffe allein oder in Kombinationen umfasst: Lenalidomid, Proteasom-Inhibitor.
- Die Patienten müssen eine vorherige Therapie mit einem monoklonalen Anti-CD38-Antikörper in einer der folgenden Formen erhalten haben
- als unmittelbar letzte Behandlung vor Studienbeginn (50 % der Patienten) oder
- Früher als während der unmittelbar letzten Behandlung vor Studienbeginn (50 % der Patienten)
Ausschlußkriterien:
- Smouldering Myelom
- Plasmazell-Leukämie
- Dokumentierte aktive systemische Amyloidose vom Leichtkettentyp.
- Multiples Myelom mit aktiver Beteiligung des Zentralnervensystems (ZNS)
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Eine zweistufige, randomisierte, multizentrische, offene Phase-3-Studie zum Vergleich von Mezigdomid (CC-92480), Bortezomib und Dexamethason (MEZIVd) mit Pomalidomid, Bortezomib und Dexamethason (PVd) bei Patienten mit rezidiviertem oder refraktärem Multiplem Myelom (RRMM): SUCCESSOR-1
Organisatorische Daten:
| Prüfplancode: | CA- 057-001 |
| ISRCTN: | |
| EudraCT: | 2021-001957-30 |
| Clinicaltrials.gov: | NCT05519085 |
| DRKS: | |
| Sponsor: | Bristol Meyer Squibb |
| Studienphase: | Phase 3 |
| Status: | Rekrutierung läuft, geplant bis Nov. 2025 |
Ziele:
Ziel der Studie ist der Vergleich der Wirksamkeit und Sicherheit von Mezigdomid (CC-92480), Bortezomib und Dexamethason (MeziVd) gegenüberPomalidomid in Kombination mit Bortezomib und Dexamethason (PVd) bei Teilnehmern mit rezidiviertem oder refraktärem Multiplem Myelom a (RRMM), die zuvor zwischen 1 bis 3 Therapielinien und bereits Lenalidomid erhalten haben.
Hintergrund:
CC-92480 ist ein sogenannter CRBN-Modulator [Cereblon-(CRBN-)E3-Ligase-Modulator (CELMoD)]. Dieser neuartige Modulator hat vielfältige Wirkungen und wirkt unter anderem stark immunmodulierend. Der Wirkstoff führt zu einem schnellen, tiefen und anhaltenden Zerfall von Ikaros und Aiolos – zwei Faktoren, die zum Überleben der Myelomzellen beitragen.
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- Der Teilnehmer hat eine dokumentierte Diagnose eines Multiplen Myeloms und eine messbare Erkrankung, gemäß der Definition im Protokoll
- Der Teilnehmer hat bereits 1 bis 3 vorherige Linien einer Myelom-Therapielinie erhalten.
- Der Teilnehmer hat ein Lenalidomid-haltiges Therapieschema erhalten.
- Der Teilnehmer hat auf mindestens eine vorherige Myelom-Therapie minimal oder besser angesprochen..
Ausschlusskriterien:
- Der Teilnehmer hat während der Behandlung oder innerhalb von 60 Tagen nach der letzten Dosis eines Proteasom-Inhibitors einen Krankheitsfortschritt (Progression) erlitten. Dies gilt nicht für Teilnehmer, die während der Behandlung mit oder innerhalb von 60 Tagen nach der letzten Dosis einer alle 2 Wochen oder seltener verabreichten Bortezomib-Erhaltungstherapie eine Progression erlitten haben, sind nicht ausgeschlossen.
- Bei Teilnehmern, die zuvor mit einem Bortezomib-haltigen Schema behandelt wurden, war das beste erreichte Ansprechen kein minimales Ansprechen (MR) oder besser, oder der Teilnehmer setzte Bortezomib aufgrund von Toxizität ab.
- Der Teilnehmer wurde zuvor mit Mezigdomid oder Pomalidomid behandelt.
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt
Subkutanes Daratumumab-Regime in Kombination mit Talquetamab oder Teclistamab zur Behandlung von Patienten mit rezidiviertem oder refraktärem Multiplem Myelom (TRIMM-2)
Organisatorische Daten:
| Prüfplancode: | |
| ISRCTN: | |
| EudraCT: | 2019-000330-19 |
| Clinicaltrials.gov: | NCT04108195 |
| DRKS: | |
| Sponsor: | Janssen |
| Studienphase: | I |
| Status: | Rekrutierung läuft, |
Ziel:
Mit dieser Studie sollen die Sicherheit von Daratumumab in Kombination mit Talquetamab oder Teclistamab sowie die vorläufige Antitumoraktivität jeder Kombination bewertet werden.
Durchführung:
Die Studie besteht aus mehreren Zeiträumen: Voruntersuchung (Screening), Behandlung (Teil 1 und 2) und Nachbeobachtung(nach Behandlungsende und bis zu 16 Wochen nach der letzten Dosis). In Teil 1 der Behandlung erhalten Patienten eine der folgenden Kombinationen in steigender Dosierung (Dosiseskalation):
- Daratumumab plus Teclistamab oder
- Daratumumab plus Talquetamab oder
- Daratumumab plus Talquetamab plus Pomalidomid oder
- Daratumumab plus Teclistamab plus Pomalidomid.
In Teil 2 der Behandlung erhalten Patienten die für die Phase 2 empfohlene Dosis, die in Teil 1 für ausgewählte Kombinationen ermittelt wurde (Dosisexpansion).
Das Studienende ist definiert als letzte Studienbeurteilung des letzten Studienteilnehmers. Die Gesamtdauer der Studie beträgt etwa 2,4 Jahre. Wirksamkeit, Sicherheit, Pharmakokinetik (PK), Immunogenität und Biomarker werden zu festgelegten Zeitpunkten bewertet. Die Sicherheit der Teilnehmer wird während der gesamten Studie überwacht.
Hintergrund:
Diese Studie beruht auf der Annahme, dass Daratumumab in Kombination mit Talquetamab oder Teclistamab aufgrund verschiedener Wirkungsmechanismen zu verbesserten klinischen Reaktionen bei der Behandlung des rezidivierten oder refraktären Multiplen Myeloms führen kann.
Daratumumab ist ein humaner monoklonaler Immunglobulin-G1-kappa-Antikörper (IgG1k), der mit hoher Affinität an CD38 (ein Oberflächenmolekül) bei einer Vielzahl von hämatologischen Malignomen einschließlich des Multiplen Myeloms bindet. Talquetamab und Teclistamab sind gegen zwei Ziele gerichtete (bispezifische) T-Zell-Redirektions-Antikörper. Talquetamab bindet an den CD3-Rezeptorkomplex auf T-Zellen und an die G-Protein-gekoppelte Rezeptorfamilie CGPRC5D, ein 7-Transmembran-Rezeptorprotein auf Plasmazellen. Teclistamab bindet an humanes und Cynomolgus-CD3- und B-Zell-Reifungsantigen (BCMA).
Einschlusskriterien:
- Dokumentierte Erstdiagnose eines Multiplen Myeloms nach den Diagnosekriterien der Internationalen Myelom-Arbeitsgruppe (IMWG)
- Einer der folgenden Punkte muss vorliegen
-
- a) mindestens 3 vorherige Therapielinien einschließlich eines Proteasom-Inhibitors (PI) (mindestens 2 Zyklen oder 2 Monate Behandlung) und eines immunmodulatorischen Medikaments (IMiD) (mindestens 2 Zyklen oder 2 Monate Behandlung) in beliebiger Reihenfolge während der Behandlung oder
- b) Krankheit, die weder auf einen PI noch ein IMiD anspricht (doppelt refraktär)
- Messbare Erkrankung beim Screening gemäß der Definition im Protokol
- ECOG (Index zur Abstufung der Lebensqualität von Patienten mit Krebserkrankungen) von 0 oder 1
Ausschlusskriterien:
- Behandlung in den vorangegangenen 3 Monaten mit einer Anti-CD38-Therapie (z.B. Daratumumab) oder Abbruch einer vorherigen Anti-CD38-Therapie zu einem beliebigen Zeitpunkt aufgrund eines unerwünschten Ereignisses im Zusammenhang mit der Anti-CD38-Therapie
- Attenuierter Lebendimpfstoff innerhalb von 4 Wochen vor der ersten Dosis des Studienmedikaments, sofern nicht vom Sponsor genehmigt
- Aktive Beteiligung des Zentralnervensystems oder klinische Anzeichen einer Hirnhautbeteiligung des Multiplen Myeloms. Bei Verdacht auf eines von beiden sind eine Magnetresonanztomographie (MRT) des Gehirns und eine Lumbal-Zytologie erforderlich.
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Eine Studie mit Talquetamab und Teclistamab jeweils in Kombination mit einem PD-1-Inhibitor (Programmed Cell Death Receptor-1) zur Behandlung von Teilnehmern mit rezidiviertem oder refraktärem Multiplem Myelom (TRIMM-3)
Organisatorische Daten:
| Prüfplancode: | CR109168, 64407564MMY1005 |
| ISRCTN: | |
| EudraCT: | 2021-005073-22 |
| Clinicaltrials.gov: | NCT05338775 |
| DRKS: | |
| Sponsor: | Janssen |
| Studienphase: | 1 |
| Status: | Rekrutierung läuft, geplant bis Sept. 2024 |
Ziel:
Ziel der Studie ist es, die sichere Dosis bzw. sicheren Dosen eines PD-1-Inhibitors in Kombination mit Talquetamab oder Teclistamab zu ermitteln und die Sicherheit und Verträglichkeit von Talquetamab oder Teclistamab bei Verabreichung in Kombination mit einem PD-1-Inhibitor zu charakterisieren.
Hintergrund:
Talquetamab ist ein humanisierter monoklonaler Antikörper, der gegen zwei Ziele gerichtet (bispezifisch) ist: gegen humanes CD3, ein Oberflächenantigen auf T-Zellen, und gegen GPRC5D, ein Antigen in Tumorzellen oder auf deren Oberfläche (tumor-assoziiertes Antigen). Bei Verabreichung bindet Talquetamab sowohl an CD3 auf T-Zellen als auch an GPRC5D, das auf bestimmten Tumorzellen wie z.B. dem Multiplen Myelom vermehrt vorkommt und eine Schlüsselrolle bei der Vermehrung von Tumorzellen spielt.
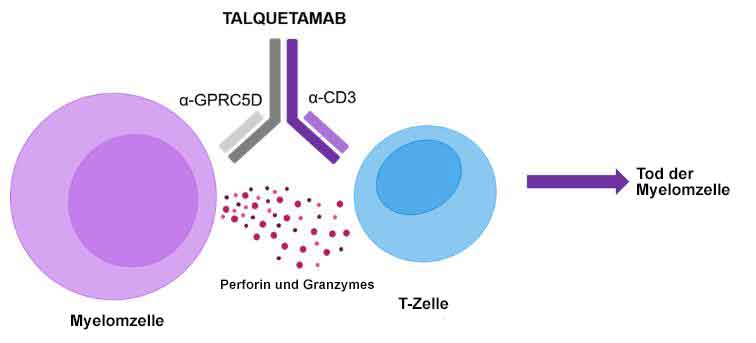
Abb. aus https://multiplemyelomahub.com/,
Teclistamab (JNJ-7957) ist ein bispezifischer Antikörper, der sowohl gegen BCMA als auch gegen CD3 gerichtet ist. BCMA ist ein Zelloberflächenprotein, das besonders auf Myelom- und Plasmazellen vorkommt und eine besondere biologische Bedeutung für das Überleben der Plasmazelle hat. Es wird bei Menschen mit Multiplem Myelom in signifikant höherem Maße exprimiert. CD3 ist an der Aktivierung der Immunantwort zur Bekämpfung von Infektionen beteiligt, Teclistamab lenkt CD3-T-Zellen auf BCMA-exprimierende Myelomzellen um, um die Zytotoxizität der Zielzellen zu induzieren. Ergebnisse aus präklinischen Studien zeigen, dass Teclistamab Myelom-Zelllinien und Myelom-Knochenmarkzellen von stark vorbehandelten Patienten abtötet.
Diese Studie geht davon aus, dass Talquetamab oder Teclistamab in Kombination mit einem PD-1-Inhibitor bei der Behandlung von rezidiviertem oder refraktärem Multiplem Myelom zu einem verbesserten klinischen Ansprechen führen, was auf verschiedene Wirkmechanismen zurückzuführen ist.
Einschlusskriterien:
- Dokumentierte Erstdiagnose eines Multiplen Myeloms nach den Diagnosekriterien der Internationalen Myelom-Arbeitsgruppe (IMWG)
- Teilnehmer mit rezidivierender oder refraktärer Erkrankung, die nicht für eine verfügbare Therapie mit nachgewiesenem klinischen Nutzen infrage kommen.
- Messbare Erkrankung zum Zeitpunkt des Screenings
- ECOG (Index zur Abstufung der Lebensqualität von Patienten mit Krebserkrankungen) von 0 oder 1
Ausschlusskriterien:
-
Vorangegangene Antitumortherapie innerhalb von 21 Tagen vor der ersten Dosis der Studienbehandlung (Therapie mit Proteasom-Inhibitoren [PI] oder Strahlentherapie innerhalb von 14 Tagen, Therapie mit immunmodulatorischen Wirkstoffen (IMiD) innerhalb von 7 Tagen, genmodifizierte adoptive Zelltherapie oder autologe Stammzelltransplantation innerhalb von 3 Monaten)
-
Vorherige Therapie mit PD-1-Inhibitoren, allogene Stammzelltransplantation oder Organtransplantation
-
Aktive Plasmazellleukämie, Waldenstrom-Makroglobulinämie, POEMS-Syndrom (Polyneuropathie, Organomegalie, Endokrinopathie, M-Protein und Hautveränderungen) oder primäre Leichtkettenamyloidose
-
Aktive Beteiligung des zentralen Nervensystems (ZNS) oder klinische Anzeichen einer meningealen Beteiligung des Multiplen Myeloms. Bei Verdacht auf eine der beiden Erkrankungen sind eine Magnetresonanztomographie des Gehirns (MRT) und eine Lumbalzytologie erforderlich.
-
Impfung mit einem abgeschwächten Lebendimpfstoff innerhalb von 4 Wochen vor der ersten Dosis der Studienbehandlung
Alle weiteren Kritereien besprechen Sie mit Ihrem Arzt
Zusätzliche Information
https://www.uke.de/kliniken-institute/kliniken/stammzelltransplantation/index.html
Unterkategorien
-
Hamburg
- COM_STUDIES_NUM
- 7
