COM_STUDIES_DEFAULT_PAGE_TITLE
Freiburg
Freiburg
Praxis für interdisziplinäre Onkologie und Hämatologie
0761-38687-0
Universitätsklinikum Freiburg, Innere Medizin I, Hämatologie/Onkologie
0761-270-0
Studien
CC-92480Ein neuer Cereblon E3 Ligase Modulator
Eine Studie zur Bestimmung der empfohlenen Dosis und des Regimes und zur Bewertung der Sicherheit und vorläufigen Wirksamkeit von CC-92480 in Kombination mit Standardbehandlungen bei Patienten mit rezidiviertem oder refraktärem Multiplen Myelom (RRMM) und neu diagnostiziertem Multiplen Myelom (NDMM)
Organisatorische Daten:
| Prüfplancode: |
CC-92480-mm-02 |
| UTMS | U1111-1233-5619 |
| EudraCT: | 2018-004767-31 |
| Clinicaltrials.gov: | NCT03989414 |
| DRKS: | |
| Sponsor: | Celgene |
| Studienphase: | Phase 1/2 |
| Status: | Rekrutierung läuft, geplant bis Januar 2025 |
Hintergrund:
CC-92480 ist ein sogenannter CRBN-Modulator [Cereblon-(CRBN-)E3-Ligase-Modulator (CELMoD)]. Dieser neuartige Modulator hat vielfältige Wirkungen und wirkt unter anderem stark immunmodulierend. Der Wirkstoff führt zu einem schnellen, tiefen und anhaltenden Zerfall von Ikaros und Aiolos – zwei Faktoren, die zum Überleben der Myelomzellen beitragen.
Durchführung:
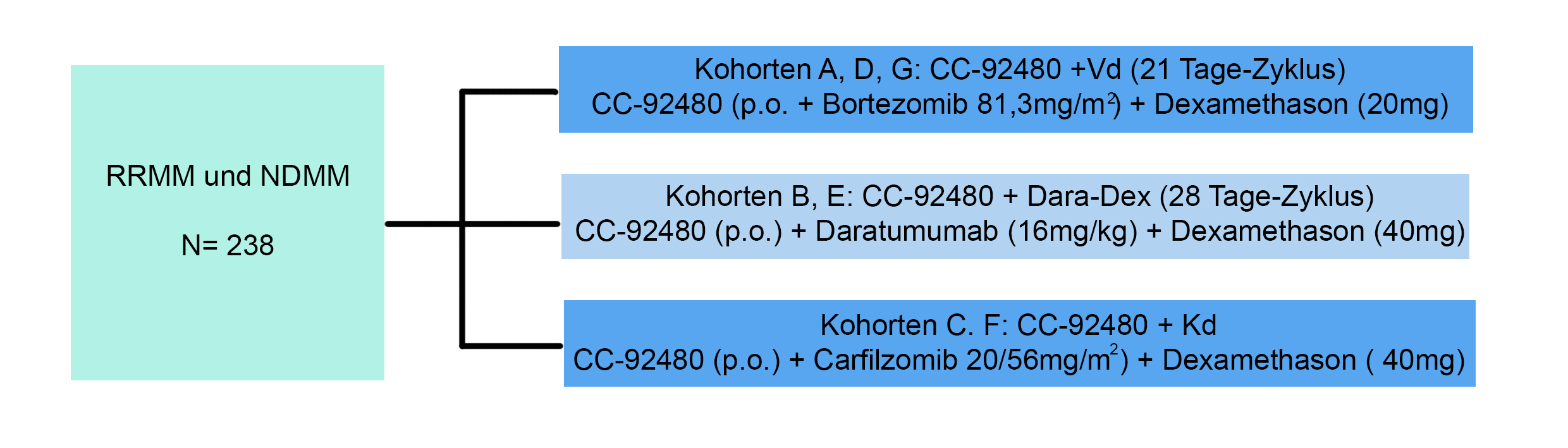
Die Studie wird randomisiert ausgeführt und die Patienten in 3 Gruppen eingeteilt, die neben dem CC-92480 unterschiedliche Zweier-Kombinationen enthalten, die gegeneinander getestet werden. Die Kohorten haben unterschiedliche Einschlusskriterien.
Ein und Ausschlusskriterien:
Einschlusskriterien:
- Alter ≥ 18 Jahre alt und ECOG (Index zur Abstufung der Lebensqualität von Patienten mit Krebserkrankungen) mit 0, 1 oder 2 Punkten.
- Patienten mit rezidiviertem oder refraktäremMultiplen Myelom müssen eine messbare Krankheit haben und ihr Krankheitsverlauf während oder nach ihrer letzten Myelomtherapie muss dokumentiert sein.
- Bei neu diagnostizierten Patienten muss die Diagnose eines zuvor unbehandelten symptomatischen Multiplen Myeloms dokumentiert sein.
- Frauen im gebärfähigen Alter und männliche Patienten müssen mit dem Schwangerschaftsverhütungsplan einverstanden sein.
Ausschlusskriterien:
- Signifikante medizinische Erkrankung, auffällige Laborwerte oder eine psychiatrische Krankheit, die an der Teilnahme an der Studie hindert.
- Patient ist nicht in der Lage oder nicht bereit, sich der laut Prüfplan vorgeschriebenen Prophylaxe einer Thromboembolie zu unterziehen.
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt
TCR-Immuntherapie mit MDG1011
Erste Phase-1/2-Studie mit TCR-Immuntherapie MDG1011 am Menschen.
Diese Dosisfindungsstudie bewertet die Sicherheit, Durchführbarkeit und vorläufige Wirksamkeit von genmodifizierten T-Zellen bei Patienten mit myeloischen und lymphatischen Neoplasien, die mit einem hohen Risiko einhergehen.
Im Phase-1-Teil dieser multizentrischen, offenen klinischen Phase-1/2-Studie sollen ca. 12 Patienten behandelt werden, die HLA-A*02:01-positiv sind und an fortgeschrittenen hämatologischen Erkrankungen wie akuter myeloischer Leukämie (AML), myelodysplastischem Syndrom (MDS) oder multiplem Myelom (MM) leiden. Die Expression von PRAME ist eine weitere Voraussetzung zur Teilnahme an dieser Dosis-Eskalations-Studie. PRAME ist ein Krebs-Antigen, welches den Zelltod und die Zelldifferenzierung hemmt und vermutlich an der malignen Entartung beteiligt ist.
In drei Dosiskohorten und einer optionalen vierten Dosiskohorte werden Dosisbereiche von 100.000 bis 10.000.000 transduzierten (gen-modifizierten) T-Zellen pro kg Körpergewicht getestet. Jede Dosiskohorte wird je einen Patienten aus jeder der drei hämatologischen Erkrankungen enthalten. Die Patienten werden eine Vorbehandlung mit Cyclophosphamid und Fludarabin erhalten. Nach der vollständigen Behandlung aller Patienten einer Dosiskohorte und einer vierwöchigen Beobachtungsperiode zur Sicherheit wird ein unabhängiges "Data Safety and Monitoring Board" (DSMB) über den Start der nächsten Dosisgruppe entscheiden.
Organisatorische Daten:
| Prüfplancode: | CD-TCR-001 |
| ISRCTN: | |
| EudraCT: | 2017-000440-18 |
| Clinicaltrials.gov: | NCT03503968 |
| DRKS: | |
| Sponsor: | Medigene AG |
| Studienphase: | 1/2 |
| Status: | Rekrutierung läuft, geplant bis August 2020 |
Ziel:
Die primären Endpunkte für den Phase-1-Teil der klinischen Studie sind Sicherheit und Verträglichkeit von MDG1011, die maximal verträgliche Dosis und die für die Phase 2 empfohlene Dosis sowie die Durchführbarkeit. Als weitere Endpunkte werden die Gesamtansprechrate, die Dauer des Ansprechens, die Zeit bis zur Krankheitsprogression, das progressionsfreie Überleben, das Gesamtüberleben, die Lebensqualität und der Zusammenhang zwischen der PRAME-Expression mit der Antitumor-Antwort nach drei Monaten beurteilt. Insgesamt beträgt die Nachbeobachtungszeit bis zu 12 Monate.
Die Endpunkte des Phase-2-Teils sind die weitere Bewertung der Sicherheit und Wirksamkeit, wobei die Wirksamkeit als Gesamtansprechrate nach 3 Monaten gemessen wird. Die Studie wird eine Nachbeobachtungszeit von bis zu 12 Monaten haben. Weitere Endpunkte umfassen die Dauer des Ansprechens, die Zeit bis zur Progression, das progressionsfreie Überleben, das Gesamtüberleben, die Lebensqualität, die Durchführbarkeit und den Zusammenhang der PRAME-Expression mit der Antitumor-Antwort.
Die experimentelle TCR-Therapie MDG1011 wird im Rahmen dieser Studie erstmals am Menschen geprüft.
Hintergrund:
Die TCR-Technologie zielt darauf ab, körpereigene T-Zellen des Patienten mit tumorspezifischen T-Zell-Rezeptoren auszustatten. Die bezüglich ihres Rezeptors modifizierten T-Zellen sind dadurch in der Lage, Tumorzellen zu erkennen und wirksam zu zerstören. Dieser immuntherapeutische Ansatz versucht, die bestehende Toleranz gegenüber den Krebszellen und die tumor-induzierte Immunsuppression im Patienten zu überwinden, indem T-Zellen des Patienten außerhalb des Körpers aktiviert und modifiziert werden.
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien (allg.):
- Dokumentierte Diagnose der Erkrankung im letzten Stadium in den letzten 4 Wochen vor dem Screening
- Humanes Leukozytenantigen (HLA)
- Phase 1 und Phase 2 (Behandlungsgruppe): Nachweis von HLA-A*02:01
- Phase 2 (Vergleichsgruppe): kein Nachweis von HLA-A*02:01
- Lebenserwartung von mindestens 4 Monaten
- ECOG (Index zur Abstufung der Lebensqualität von Patienten mit Krebserkrankungen) von 0 bis 2
- keine geplante allogene hämatopoetische Stammzelltransplantation (HSZT)
Multiples Myelom-(MM-)spezifische Einschlusskriterien:
- Rezidiviertes und refraktäres MM
- Mindestens 3 vorangegangene Therapielinien mit mindestens einem Proteasomeninhibitor und einer immunmodulierenden Substanz (IMiD). Eine Induktion mit oder ohne hämatopoetische Stammzelltransplantation und mit oder ohne Erhaltungstherapie wird als ein einziges Behandlungsschema betrachtet.
- Myelomzellen müssen PRAME positiv exprimieren
Ausschlußkriterien:
- Akute Promyelozyten-Leukämiamit t(15;17)(q22;q12); Promyelozyten-Leukämia/Retinoic Acid Receptor Alpha (PML-RARA) oder mit varianten Translokationen
- Bekannter Nachweis des HIV-Virus, aktive Hepatitis-B-Virus-(HBV-) oder Hepatitis-C-Virus-(HCV-)Infektion
- Klinisch signifikante, fortgeschrittene oder instabile Erkrankung oder unzureichende Funktion eines Hauptorgans, die den Teilnehmer einem besonderen Risiko aussetzen könnte
- allogene Stammzelltransplantation in der Vorgeschichte
MM-spezifische Ausschlusskriterien für Phase 1 und Phase 2 (Behandlungsgruppe, falls das MM in Phase 2 fortschreitet):
- Vortherapie mit immunmodulierenden Substanzen (IMiDs) innerhalb von 14 Tagen vor der Leukapherese und/oder der Infusion des Prüfpräparats
- Vortherapie mit Kortikosteroiden innerhalb von 7 Tagen vor der Leukapherese oder 7 Tagen vor der Infusion des Prüfpräparats
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Teclistamab, Daratumumab, Lenalidomid,Dexamethason +/- Bortezomib
Phase-2-Studie zur Bewertung der Sicherheit und Wirksamkeit von Teclistamab in Kombination mit Daratumumab, Lenalidomid und Dexamethason mit und ohne Bortezomib als Induktionstherapie und Teclistamab in Kombination mit Daratumumab und Lenalidomid als Erhaltungstherapie bei transplantationsgeeigneten Patienten mit neu diagnostiziertem Multiplem Myelom
Organisatorische Daten:
| Prüfplancode: | GMMG-HD10, DSMM-XX, 64007957MMY2003, MajesTEC-5 |
| ISRCTN: | |
| EudraCT: | |
| Clinicaltrials.gov: | NCT05695508 |
| DRKS: | |
| Sponsor: | Universitätsklinikum Heidelberg in Zusammenarbeit mit Janssen |
| Studienphase: | Phase 3 |
| Status: | Rekrutierung läuft, geplant bis Mai 2026 |
Hintergrund:
Teclistamab ist ein bispezifischer Antikörper, der an das B-Zell-Reifungsantigen (BCMA), ein Protein auf der Oberfläche von Myelomzellen, bindet und gegen CD3-Rezeptoren auf der Oberfläche von T-Zellen gerichtet ist. Teclistamab wurde im August 2022 in der EU als Monotherapie zur Behandlung des fortgeschrittenen Multiplen Myeloms zugelassen und wird jetzt bei weiteren Indikationen geprüft.
In dieser Studie bei Patienten mit neu diagnostiziertem Multiplem Myelom, die für eine Transplantation geeignet sind, werden die Sicherheit, Verträglichkeit und Wirksamkeit einer Fünffach-Kombination in der Induktionstherapie geprüft: Teclistamab (Tec) in Kombination mit Daratumumab (Dara), Lenalidomid (R), Bortezomib (V) und Dexamethason (d).
Durchführung
Das Studiendesign gliedert sich in drei Studienarme:
Patienten in Arm A und B erhalten eine Induktionstherapie mit Tec-Dara-Rd (Arm A) oder Tec-Dara-VRd (Arm B). Es folgen eine Hochdosis-Chemotherapie und eine autologe Stammzelltransplantation gemäß Therapiestandard sowie eine Erhaltungstherapie mit Tec-Dara-R.
Patienten in Arm C erhalten eine Induktionstherapie gemäß Therapiestandard gefolgt von einer Hochdosis-Chemotherapie und einer autologen Stammzelltransplantation sowie gegebenenfalls einer Konsolidierungstherapie. Im Anschluss daran folgt eine Erhaltungstherapie mit Tec-Dara-R.
Die Behandlungsdauer der Induktionsphase beträgt 6 Zyklen (à 28 Tage). Im Anschluss daran folgen eine Hochdosis-Chemotherapie und autologe Stammzelltransplantation . Danach beginnt die Erhaltungsphase der Studie unter Tec-Dara-R über 18 Zyklen.
Studienarm C beginnt mit dem Screening nach abgeschlossener Hochdosis-Chemotherapieplus autologer Stammzelltransplantation. Die Behandlung erfolgt genauso wie in Arm A und B mit Tec-Dara-R als Erhaltungstherapie für 18 Zyklen.
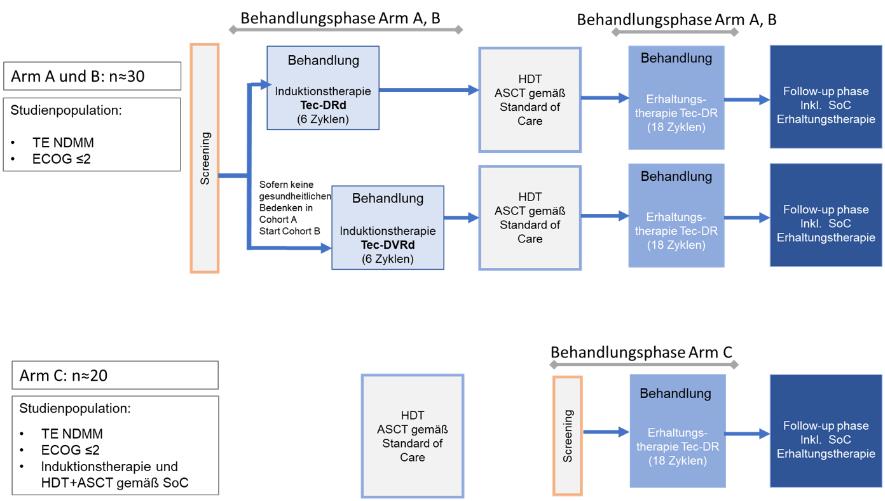
Abb. Studiendesign, mit freundlicher Genehmigung des Universitätsklinikums Heidelberg, Prof. Raab
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- Neu diagnostiziertes Multiples Myelom gemäß den Diagnosekriterien der International Myeloma Working Group (IMWG) und Erhalt einer Induktionstherapie mit oder ohne Konsolidierung
- Ein ECOG-Performance-Status-Score (Index zur Abstufung der Lebensqualität von Patienten mit Krebserkrankungen) von 0, 1 oder 2 beim Screening und unmittelbar vor Beginn der Verabreichung der Studienbehandlung
- Messbare Erkrankung während der Screening-Phase, gemäß der Definition im Protokoll
Ausschlußkriterien
- Beteiligung des ZNS oder klinische Anzeichen für eine Beteiligung der Hirnhäute
- Schlaganfall oder Krampfanfall innerhalb von 6 Monaten vor Studienbeginn
- Transplantation in der Krankengeschichte, die eine immunsuppressive Behandlung erforderte
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Eine offene, multizentrische Studie der Phase 1/2 zur Sicherheit, Verträglichkeit und Pharmakokinetik von HPN217 bei Patienten mit rezidiviertem /refraktärem Multiplem Myelom.
Organisatorische Daten:
| Prüfplancode: | HPN 217-3001 |
| ISRCTN: | |
| EudraCT: | |
| Clinicaltrials.gov: | NCT04184050 |
| DRKS: | |
| Sponsor: | Harpoon Therapeutics |
| Studienphase: | 1/2 Erstanwendung Menschen |
| Status: | Rekrutierung läuft, geplant bis Januar 2024 |
Primäres Ziel:
Diese Erstanwendung am Menschen (First-in-Human-Studie) der Phase 1 wird in zwei Teilen durchgeführt. Zweck der Dosissteigerung (Eskalation) in Teil 1 ist es festzustellen, ob HPN 217, das als wöchentliche Kurzzeit-Infusionen verabreicht wird, sicher und verträglich ist. Danach folgt Teil 2 mit einer Dosiserweiterung (Expansion), um weitere Wirksamkeits- und Sicherheitserfahrungen mit bei erwachsenen Probanden mit rezidiviertem / refraktärem multiplem Myelom zu sammeln. Die Sicherheit der Probanden wird durch intensive Vitalparameteruntersuchungen, Elektrokardiogramme, körperliche Untersuchungen und Laboruntersuchungen überwacht.
Sekundäres Ziel:
Beurteilung der Sicherheit und Machbarkeit von HPN 2017, sowie der Wirksamkeit, Sicherheit.
Hintergrund:
HPN217, das aus dem Tri-spezifischem T-Zell-Aktivierungskonstrukt („TriTAC“) des Unternehmens Harpoon erzeugt wird, ist so konstruiert, dass es die patienteneigenen T-Zellen so umlenkt, dass sie BCMA-positive (engl. B-cell maturation antigen; B-Zell-Reifungsantigen) Krebszellen abtöten. HPN217 ist das erste TriTAC von Harpoon, das für die Behandlung von hämatologischen Krebsarten wie dem Multiplen Myelom entwickelt wurde. Die Daten wurden auf der Jahrestagung der American Society of Hematology (ASH) 2018 in San Diego vom 1. bis 4. Dezember vorgestellt.
Einschlusskriterien:
Multiples Myelom, das die folgenden Kriterien erfüllt:
- Mindestens 3 Vortherapien (einschließlich Proteasom-Inhibitor, immunmodulierendes Medikament und ein Anti-CD38-Antikörper; Patienten sollten nicht für eine der etablierten Therapien, die bekanntlich einen klinischen Nutzen beim Multiplen Myelom bieten. geeignet sein oder weisen ein Unverträglichkeit gegenüber all diesen Therapie auf).
- Messbare Krankheit mit mindestens folgenden Laborparametern:
- Serum M-Protein ≥ 0,5 g/dl
- Urin M-Protein ≥ 200 mg/24 Stunden
- freie Leichtketten im Serum (FLC) ≥ 10 mg/lL (≥ 100 mg/l)
- ECOG Performance Status (Index zur Abstufung der Lebensqualität von Patienten mit Krebserkrankungen) von ≤ 2
Ausschlusskriterien
- Vorhergehende Behandlung mit Substanzen, die gegen BCMA gerichtet sind (nur in Teil 2 der Studie)
- Gleichzeitige Behandlung mit Antitumornekrosefaktor-Alpha-Therapien, systemischen Kortikoidsteroiden (Prednison-Dosis > 10 mg pro Tag oder gleichwertige Dosis) oder anderen immunsuppressiven Medikamenten innerhalb der 2 Wochen vor dem Screening
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Subkutanes Daratumumab-Regime in Kombination mit Talquetamab oder Teclistamab zur Behandlung von Patienten mit rezidiviertem oder refraktärem Multiplem Myelom (TRIMM-2)
Organisatorische Daten:
| Prüfplancode: | |
| ISRCTN: | |
| EudraCT: | 2019-000330-19 |
| Clinicaltrials.gov: | NCT04108195 |
| DRKS: | |
| Sponsor: | Janssen |
| Studienphase: | I |
| Status: | Rekrutierung läuft, |
Ziel:
Mit dieser Studie sollen die Sicherheit von Daratumumab in Kombination mit Talquetamab oder Teclistamab sowie die vorläufige Antitumoraktivität jeder Kombination bewertet werden.
Durchführung:
Die Studie besteht aus mehreren Zeiträumen: Voruntersuchung (Screening), Behandlung (Teil 1 und 2) und Nachbeobachtung(nach Behandlungsende und bis zu 16 Wochen nach der letzten Dosis). In Teil 1 der Behandlung erhalten Patienten eine der folgenden Kombinationen in steigender Dosierung (Dosiseskalation):
- Daratumumab plus Teclistamab oder
- Daratumumab plus Talquetamab oder
- Daratumumab plus Talquetamab plus Pomalidomid oder
- Daratumumab plus Teclistamab plus Pomalidomid.
In Teil 2 der Behandlung erhalten Patienten die für die Phase 2 empfohlene Dosis, die in Teil 1 für ausgewählte Kombinationen ermittelt wurde (Dosisexpansion).
Das Studienende ist definiert als letzte Studienbeurteilung des letzten Studienteilnehmers. Die Gesamtdauer der Studie beträgt etwa 2,4 Jahre. Wirksamkeit, Sicherheit, Pharmakokinetik (PK), Immunogenität und Biomarker werden zu festgelegten Zeitpunkten bewertet. Die Sicherheit der Teilnehmer wird während der gesamten Studie überwacht.
Hintergrund:
Diese Studie beruht auf der Annahme, dass Daratumumab in Kombination mit Talquetamab oder Teclistamab aufgrund verschiedener Wirkungsmechanismen zu verbesserten klinischen Reaktionen bei der Behandlung des rezidivierten oder refraktären Multiplen Myeloms führen kann.
Daratumumab ist ein humaner monoklonaler Immunglobulin-G1-kappa-Antikörper (IgG1k), der mit hoher Affinität an CD38 (ein Oberflächenmolekül) bei einer Vielzahl von hämatologischen Malignomen einschließlich des Multiplen Myeloms bindet. Talquetamab und Teclistamab sind gegen zwei Ziele gerichtete (bispezifische) T-Zell-Redirektions-Antikörper. Talquetamab bindet an den CD3-Rezeptorkomplex auf T-Zellen und an die G-Protein-gekoppelte Rezeptorfamilie CGPRC5D, ein 7-Transmembran-Rezeptorprotein auf Plasmazellen. Teclistamab bindet an humanes und Cynomolgus-CD3- und B-Zell-Reifungsantigen (BCMA).
Einschlusskriterien:
- Dokumentierte Erstdiagnose eines Multiplen Myeloms nach den Diagnosekriterien der Internationalen Myelom-Arbeitsgruppe (IMWG)
- Einer der folgenden Punkte muss vorliegen
-
- a) mindestens 3 vorherige Therapielinien einschließlich eines Proteasom-Inhibitors (PI) (mindestens 2 Zyklen oder 2 Monate Behandlung) und eines immunmodulatorischen Medikaments (IMiD) (mindestens 2 Zyklen oder 2 Monate Behandlung) in beliebiger Reihenfolge während der Behandlung oder
- b) Krankheit, die weder auf einen PI noch ein IMiD anspricht (doppelt refraktär)
- Messbare Erkrankung beim Screening gemäß der Definition im Protokol
- ECOG (Index zur Abstufung der Lebensqualität von Patienten mit Krebserkrankungen) von 0 oder 1
Ausschlusskriterien:
- Behandlung in den vorangegangenen 3 Monaten mit einer Anti-CD38-Therapie (z.B. Daratumumab) oder Abbruch einer vorherigen Anti-CD38-Therapie zu einem beliebigen Zeitpunkt aufgrund eines unerwünschten Ereignisses im Zusammenhang mit der Anti-CD38-Therapie
- Attenuierter Lebendimpfstoff innerhalb von 4 Wochen vor der ersten Dosis des Studienmedikaments, sofern nicht vom Sponsor genehmigt
- Aktive Beteiligung des Zentralnervensystems oder klinische Anzeichen einer Hirnhautbeteiligung des Multiplen Myeloms. Bei Verdacht auf eines von beiden sind eine Magnetresonanztomographie (MRT) des Gehirns und eine Lumbal-Zytologie erforderlich.
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Venetoclax in Kombination mit Daratumumab /Dexamethason
Eine Studie zur Kombinationstherapie mit Venetoclax, Daratumumab und Dexamethason (mit und ohne Bortezomib) bei Patienten mit rezidiviertem oder refraktärem Multiplem Myelom.
Organisatorische Daten:
| Prüfplancode: | M15-654 |
| ISRCTN: | |
| EudraCT: | 2017-002099-26 |
| Clinicaltrials.gov: | NCT03314181 |
| DRKS: | |
| Sponsor: | AbbVie in Zusammenarbeit mit Janssen Research & Development, LLC |
| Studienphase: | Phase 1/2 |
| Status: | Rekrutierung läuft, geplant bis 6/2024 |
Kurzbeschreibung:
Multizentrische Dosissteigerungs- und Erweiterungsstudie der Phase 1 / 2 zur Bewertung der Sicherheit, Verträglichkeit und Wirksamkeit der Kombinationstherapie mit Venetoclax, Daratumumab und Dexamethason mit und ohne Bortezomib bei Teilnehmern mit rezidiviertem oder refraktärem multiplen Myelom zu bewerten.
Ziel:
In Teil 1 dieser Studie soll die objektive Ansprechrate in einem Zeitraum von bis zu 3 Monaten und die Anzahl der Teilnehmer mit dosisbegrenzenden Nebenwirkungen (Toxizitäten) ermittelt werden.
In Teil 2 dieser Studie soll die Anzahl der Teilnehmer mit dosisbegrenzenden Nebenwirkungen und das vollständige Ansprechen in einem Zeitraum von bis zu 5 Monaten ermittelt werden.
Hintergrund:
Venetoclax ist ein BCL-2-Inhibitor, der zur Behandlung der chronisch lymphatischen Leukämie bereits zugelassen ist. Es zählt chemisch gesehen zu den kleinen Molekülen, auch small molecules" genannt.
Venetoclax hemmt selektiv das Protein Bcl-2 das bei verschiedenen Krebsarten, einschließlich Multiples Myelom vermehrt vorkommt. Bcl-2 unterdrückt den programmierten Zelltod (Apoptose) der Tumorzellen. Venetoclax hingegen blockiert Bcl-2 im Zellinneren und führt so zum programmierten Zelltod der Tumorzellen. Dadurch wird auch die Wirksamkeit herkömmlicher Chemotherapeutika verbessert.
Einschlusskriterien:
- ECOG (Index zur Abstufung der Lebensqualität von Patienten mit Krebserkrankungen) von 0, 1 oder 2
- Rezidiviertes oder refraktäres Multiples Myelom mit dokumentierter Progression, die während oder nach dem letzten Behandlungsschema aufgetreten ist und vom Prüfer anhand der Kriterien der International Myeloma Working Group (IMWG) bestimmt wurde
- anhand, muss der Teilnehmer t(11;14) positiv sein [nachgewiesen in einem Zentrallabor durch einen sogenannten analytisch validierten Fluoreszenz-in-Situ-Hybridisierungs-(FISH)-Assay]
Ausschlusskriterien:
- Frühere Behandlung mit Venetoclax oder einem anderen B-Zell-Lymphoma-2-(BCL-2-) Inhibitor ODER frühere Behandlung mit Daratumumab oder einer anderen Anti-CD38-Therapie.
- Für Teilnehmer von Teil 2:
- Der Teilnehmer ist gegenüber einem Proteasom-Inhibitor refraktär, was definiert ist als Progression an oder innerhalb von 60 Tagen nach der letzten Dosis eines Behandlungsschemas mit einem Proteasominhibitor
- Behandlung mit einem Proteasom-Inhibitor innerhalb von 60 Tagen vor der ersten Dosis des Studienmedikaments
- Behandlung mit Antimyelom-Chemotherapie, Strahlentherapie, biologischer Therapie, Immuntherapie oder einer Forschungstherapie, einschließlich gezielter niedermolekularer Wirkstoffe innerhalb von 2 Wochen oder gegebenenfalls 5 Halbwertszeiten vor der ersten Dosis.
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt
