COM_STUDIES_DEFAULT_PAGE_TITLE
Berlin
Berlin
Charité - Universitätsmedizin Berlin der Freien Universität und der Humboldt-Universität, Campus Benjamin Franklin
030-4505-0
Studien
AlloRelapseMM
Allogene Stammzelltransplantation im Vergleich zur konventionellen Therapie als Salvage-Therapie für Patienten mit rezidiviertem / progredientem Multiplen Myelom nach einer Erstlinientherapie (AlloRelapseMMStudy)
Organisatorische Daten:
| Prüfplancode: | |
| ISRCTN: | |
| EudraCT: | 2021-001--5-67 |
| Clinicaltrials.gov: | NCT0567319 |
| DRKS: | |
| Sponsor: | Universitätsklinikum Hamburg-Eppendorf mit Unterstützung des Gemeinsamen Bundesausschusses (G-BA) |
| Studienphase: | III |
| Status: | Rekrutierung läuft, geplant bis 6/2027 |
Ziel:
In dieser Studie wird untersucht, ob die Transplantation von gespendeten Stammzellen (allogene Stammzelltransplantation) Vorteile gegenüber der gängigen Therapie bei Patienten mit Multiplem Myelom bietet, bei denen es nach einer Transplantation von körpereigenen Stammzellen (autologe Stammzelltransplantation) als Erstlinientherapie zu einem Erkrankungsrückfall (Rezidiv) oder einem Fortschreiten der Krankheit (Progression) gekommen ist. Es wird geprüft, ob die allogene Stammzelltransplantation das Gesamtüberleben bei diesen Patienten verlängert.
Hintergrund:
Eine Behandlungsoption des Multiplen Myeloms ist die Stammzelltransplantation. Nach einer Chemotherapie, bei der die Krebszellen zerstört werden, werden körpereigene (autologe) oder gespendete körperfremde (allogene) Stammzellen übertragen. Die übertragenen Stammzellen siedeln sich dann im Knochenmark an und bilden „frische“ Blutzellen. Die Stammzelltransplantation kommt als Behandlungsoption nach der ersten Diagnose (Erstlinientherapie), aber auch bei einem Rückfall nach Vorbehandlung (jenseits der Erstlinientherapie) in Frage. Auch wenn das Multiple Myelom oft nicht endgültig heilbar ist, soll die Therapie die Lebenserwartung verlängern und die Symptome zurückdrängen. (Quelle: https://www.g-ba.de/studien/erprobung/allorelapsemmstudy-studie/)
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- Diagnose Multiples Myelom,
- Rückfall (Rezidiv) oder Fortschreiten (Progress) des Multiplen Myeloms unter oder nach einer autologen Stammzelltransplantation
- Erhalt von höchstens 1 Zyklus einer sogenannten Salvage-Therapie (Rezidiv-Therapie) vor Einschluss in die Studie
Ausschlusskriterien:
- Unzureichende Organfunktion gemäß der Definition im Prüfplan
- Aktive Hepatitis B oder C oder unkontrollierte HIV-Infektion
- Andere aktive bösartige Erkrankung
- Vorherige Behandlung mit allogenen Stammzellen
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Offene Phase-I/II-Multikohortenstudie zur Bewertung der Wirksamkeit und Sicherheit von Cevostamab bei Patienten mit rezidiviertem/refraktärem Multiplem Myelom, die zuvor ein Anti-B-Zell-Reifungsantigen erhalten haben
Organisatorische Daten:
| Prüfplancode: | CO43476 |
| ISRCTN: | |
| EudraCT: | 2021-006816-10 |
| Clinicaltrials.gov: | NCT05535224 |
| DRKS: | |
| Sponsor: | Hoffmann La Roche |
| Studienphase: | Phase 1 |
| Status: | Rekrutierung geplant bis Febr. 2027 |
Hintergrund:
Cevostamab ist ein neuer, gegen zwei Ziele gerichteter (bispezifischer) Antikörper. Er zielt auf einen Rezeptor, der ausschließlich auf Zellen der B-Linie exprimiert wird. Bei stark vorbehandelten Patienten mit rezidiviertem/refraktärem Multiplem Myelom (RRMM) hat Cevostamab eine vielversprechende Wirkung gezeigt. In dieser Studie soll die Wirksamkeit und Sicherheit von Cevostamab als mögliche Therapie für Multiples Myelom weiter untersucht werden. Die Studie wird in unterschiedlichen Kohorten durchgeführt.
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- Dokumentierte Diagnose eines Multiplen Myeloms auf der Grundlage der Standardkriterien der International Myeloma Working Group (IMWG)
- Nachweis eines Fortschreitens der Erkrankung auf der Grundlage der Bestimmung des Ansprechens durch die Prüfer nach den IMWG-Kriterien bei oder nach der letzten Dosierung
- Vorangegangene BCMA-ADC- oder CAR-T-Kohorte: Teilnehmer, die eine gegen BCMA gerichtete Therapie mit Chimären Antigenrezeptor-T-Zellen (CAR-T-Zellen) oder Antikörper-Wirkstoff-Konjugat (ADC)) erhalten haben und auf eine Dreifachtherapie nicht oder nicht mehr ansprechen (refraktär sind)
- Vorangegangene BCMA Bispezifische Kohorte: Teilnehmer, die einen gegen BCMA gerichteten T-Zell-abhängigen bispezifischen (TDB) Antikörper erhalten haben und auf eine Dreifachtherapie nicht oder nicht mehr ansprechen (refraktär sind)
- Messbare Krankheit gemäß den Diagnosekriterien der International Myeloma Working Group (IMWG)
- ECOG- Leistungsstatus (Index zur Abstufung der Lebensqualität von Patienten mit Krebserkrankungen) von 0 oder 1
Ausschlußkriterien:
- Unfähigkeit, den nach dem Protokoll vorgeschriebenen Krankenhausaufenthalt einzuhalten
- Schwangerschaft oder Stillen oder die Absicht, während der Studie oder innerhalb von 5 Monaten nach der letzten Dosis von Cevostamab oder Tocilizumab oder innerhalb von 3 Monaten nach der letzten Dosis von Tocilizumab schwanger zu werden (falls zutreffend)
- Vorherige Behandlung mit Cevostamab oder einem anderen Wirkstoff, der gegen dasselbe Ziel gerichtet ist
- Vorherige BCMA-ADC- oder CAR-T-Kohorte: vorherige Behandlung mit einem beliebigen T-Zell-abhängigen bispezifischen Antikörper (TDB-Antikörper), einschließlich TDB, die nicht auf BCMA abzielen
- Vorherige Anwendung eines monoklonalen Antikörpers, eines Radioimmunkonjugats oder eines ADC als Krebstherapie innerhalb von 4 Wochen vor der ersten Studienbehandlung, ausgenommen die Anwendung einer Nicht-Myelom-Therapie
- Vorherige Behandlung mit systemischen Immuntherapeutika
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Induktionstherapie mit Lenalidomid, Bortezomib und Dexamethason und entweder intravenösem oder subkutanem Isatuximab bei Patienten mit neu diagnostiziertem Multiplem Myelom, die für eine Hochdosis-Chemotherapie mit nachfolgender autologer Stammzelltransplantation geeignet sind
Organisatorische Daten:
| Prüfplancode: | GMMG-HD 8 / DSMM XIX |
| ISRCTN: | |
| EudraCT: | 2022-000996-38 |
| Clinicaltrials.gov: | NCT05804032 |
| DRKS: | |
| Sponsor: | Universitätsklinikum Heidelberg DSMM, Sanofi und KKS-Netzwerk |
| Studienphase: | |
| Status: | Rekrutierung geplant bis Juni 2025 |
Ziel:
In dieser Studie für Patienten mit zuvor unbehandeltem Multiplem Myelom soll die Wirksamkeit der Induktionstherapie mit Isatuximab in Kombination mit Lenalidomid/Bortezomib/Dexamethason (RVd) bei Gabe von Isatuximab unter die Haut (subkutan) gegenüber einer Gabe in die Vene (intravenös) untersucht werden.
Durchführung:
Randomisierung: Die Patienten werden vor der Induktionstherapie nach dem Zufallsverfahren (randomisiert) einem von 2 Studienarmen (A oder B) zugeordnet.
- Patienten in Arm A erhalten 3 Zyklen des monoklonalen Antikörpers Isatuximab intravenös in Kombination mit dem RVd-Schema (Lenalidomid, Bortezomib, Dexamethason). Jeder Zyklus hat eine Dauer von 42 Tagen.
- Patienten in Arm B erhalten 3 Zyklen Isatuximab subkutan in Kombination mit RVd.
Nach der Induktionstherapie erhalten die Patienten eine Standard-Intensivierung (in der Regel eine Mobilisierungstherapie auf Cyclophosphamid-Basis, Stammzellenentnahme und hochdosiertes Melphalan, gefolgt von einer autologen Stammzelltransplantation
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- Bestätigte Diagnose eines neu diagnostizierten behandlungsbedürftigen Multiplen Myeloms gemäß den überarbeiteten Diagnosekriterien der International Myeloma Working Group (IMWG))
- Der Patient ist für eine Hochdosis-Chemotherapie und eine autologe Stammzelltransplantation geeignet
- Messbare Krankheitsaktivität (gemäß der Definition im Prüfplan)
- Alter 18 bis 70 Jahre bei Studieneinschluss
Ausschlußkriterien:
- Bekannte Überempfindlichkeit (oder Kontraindikation) gegen einen der Bestandteile der Studientherapie
- Systemische Amyloid-Leichtketten-Amyloidose (außer lokalisierte Amyloid-Leichtketten -Amyloidose, die auf die Haut oder das Knochenmark beschränkt ist)
- Plasmazell-Leukämie
- Vorherige Chemo- oder Strahlentherapie in den letzten 5 Jahren (außer lokaler Strahlentherapie bei lokalem Fortschreiten des Myeloms)
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Eine Phase-I-Studie mit MAGE-A1-spezifischen TCR-veränderten T-Zellen bei Patienten mit rezidiviertem/refraktärem multiplem Myelom
Organisatorische Daten:
| Prüfplancode: | MAGE-A1-TCR |
| ISRCTN: | |
| EudraCT: | 2017-001208-30 |
| Clinicaltrials.gov: | |
| DRKS: | DRKS00020221 |
| Sponsor: | Charité Campus Berlin Buch mit Förderung des Bundesministeriums für Bildung und Forschung |
| Studienphase: | Phase 1 |
| Status: | Rekrutierung läuft |
Ziel:
Untersuchung der Sicherheit und Verträglichkeit ansteigender Einzeldosen von genetisch veränderten, MAGE-A1-spezifischen, zytotoxischen autologen T-Zellen bei Patienten mit rezidiviertem und/oder refraktärem multiplem Myelom
Hintergrund:
T1367 T-Zellen sind autologe, zytotoxische (CD8+) T-Zellen, welche aus dem Blut entnommen und dann im Labor genetisch so verändert werden, dass sie gezielt Myelom-Zellen erkennen und zerstören können.
Durchführung
Die Studie besteht aus mehreren Phasen
1. Voruntersuchung
2. Vorlaufphase, in der T-Zellen mit einer Leukapherese aus dem Blut gefiltert und zu T1367 T-Zellen verändert werden (Dauer ca. 6 Wochen) sowie einer optionalen Überbrückungstherapie bei Patienten mit sehr aggressiver bzw. schnell fortschreitender Erkrankung,
3. Behandlungsphase mit Chemotherapie (Cyclophosphamid und Fludarabin) über 3 Tage und anschließender einmaliger intravenöser Verabreichung der T1367 T-Zellen sowie einer Nachuntersuchung. Während der Behandlungsphase sind die Patienten ca. 20 Tage stationär aufgenommen und werden engmaschig überwacht.
Es werden 3-6 Patienten pro Dosierungsstufe eingeschlossen. Jeder Patient wird nur in jeweils einer Dosisstufe behandelt.
Die vorgesehenen Dosierungsstufen sind 100.000, 1 Million, 10 Millionen und 50 bis 100 Millionen T-Zellen pro kg Körpergewicht (jeweils ± 20%).
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- Dokumentierte Diagnose eines rezidivierten/ refraktären multiplen Myeloms
- ECOG Performance Status von 0 bis 2 ((Index zur Abstufung der Lebensqualität von Patienten mit Krebserkrankungen)
- Mindestens 3 vorherige Therapielinien
- Nachgewiesene Expression von MAGE-A1 in mindestens 30% der Tumorzellen in einer Biopsie des Knochenmarks oder einer Manifestation außerhalb des Knochenmarks
Ausschlusskriterien:
- Allogene Stammzelltransplantation
- Jedwede Behandlung mit einer Gentherapie oder gentechnisch veränderten Immuntherapie
- Aktive oder chronische Hepatitis
- HIV Infektion
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Studie zu Elranatamab als Monotherapie und Elranatamab + Daratumumab im Vergleich zu Daratumumab + Pomalidomid + Dexamethason bei Teilnehmern mit rezidiviertem/refraktärem Multiplem Myelom
Organisatorische Daten:
| Prüfplancode: | C1071005, MagnetisMM-5 |
| ISRCTN: | |
| EudraCT: | 2021-000044-22 |
| Clinicaltrials.gov: | NCT05020236 |
| DRKS: | |
| Sponsor: | Pfizer |
| Studienphase: | Phase 3 |
| Status: | Rekrutierung läuft, geplant bis Okt. 2024 |
Ziel:
Mit dieser Studie soll untersucht werden, ob der bispezifische BCMA/CD3-Antikörper Elranatamab allein und/oder in Kombination mit dem monoklonalen Anti-CD38-Antikörper Daratumumab für Patienten mit Multiplem Myelom einen größeren Nutzen bringt als eine Kombinationstherapie aus Daratumumab, Pomalidomid und Dexamethason.
Hintergrund:
Der bispezifische BCMA/CD3-Antikörper Elranatamab (PF-06863135) zeigte bei Patienten mit rezidiviertem/refraktärem Multiplem Myelom eine vielversprechende vorläufige Wirksamkeit und Verträglichkeit.
Daten aus der Phase-1-Studie MagnetisMM-1 (NCT03269136), die auf der ASCO-Jahrestagung 2021 vorgestellt wurden, zeigten, dass Elranatamab in Dosen von bis zu 1000 µg/kg ein überschaubares Sicherheitsprofil ohne dosislimitierende Toxizitäten aufweist.
Darüber hinaus erzielte der Wirkstoff bei Dosierungen von 215 µg/kg und höher eine Gesamtansprechrate (ORR) von 70 % und eine vollständige Ansprechrate (CR)/stringente CR von 30 %. Bei der empfohlenen Phase-2-Dosis von 1000 µg/kg erzielte der Wirkstoff eine ORR von 83,3 %. Bemerkenswert ist, dass drei von vier in die Studie aufgenommenen Patienten, die zuvor mit einer BCMA-gerichteten Therapie behandelt worden waren, ein Ansprechen erzielten.
Durchführung:
In die Studie werden Patienten mit Multiplem Myelom aufgenommen, die zuvor mit Lenalidomid und einem Proteasom-Inhibitor behandelt wurden.
In Teil 1 der Studie wird die Sicherheit und Wirksamkeit verschiedener Dosen von Elranatamab in Kombination mit Daratumumab untersucht.
Die Teilnehmer an Teil 2 der Studie werden nach dem Zufallsprinzip einer von drei Behandlungsgruppen zugeteilt.
- Teilnehmer in Gruppe 1 erhalten Elranatamab allein als Injektion unter die Haut.
- Teilnehmer der Gruppe 2 erhalten Elranatamab und Daratumumab als Injektion unter die Haut.
- Teilnehmer der Gruppe 3 erhalten die Standardtherapie Daratumumab als Injektion unter die Haut sowie Pomalidomid und Dexamethason zur Einnahme.
In Teil 2 wird die Sicherheit und Aktivität von (1) Elranatamab allein im Vergleich zu Daratumumab, Pomalidomid und Dexamethason und (2) Elranatamab plus Daratumumab im Vergleich zu Daratumumab, Pomalidomid und Dexamethason verglichen. Die Teilnehmer beider Studienteile erhalten die Studienbehandlung so lange, bis ihre Krankheit fortschreitet, unannehmbare Nebenwirkungen auftreten oder sie sich entscheiden, nicht weiter an der Studie teilzunehmen.
Einschlusskriterien:
- Diagnose eines Multiplen Myeloms gemäß den von der International Myeloma Working Group (IMWG) aufgestellten Diagnosekriterien )
- Messbare Erkrankung, definiert durch mindestens 1 der folgenden Punkte:
- M-Protein im Serum > 0,5 g/dl durch SPEP
- M-Protein-Ausscheidung im Urin > 200 mg/24 Stunden mittels UPEP
- Freie Immunglobulin-Leichtketten (FLC) im Serum ≥ 10 mg/dl (≥ 100 mg/l) UND abnormes Verhältnis der Immunglobuline Kappa-zu-Lambda-FLC-im Serum
- Vorherige Therapie gegen multiples Myelom, einschließlich Behandlung mit Lenalidomid und einem Proteasom-Inhibitor.
- ECOG Performance Status (Index zur Abstufung der Lebensqualität von Patienten mit Krebserkrankungen) von 0, 1 oder 2.
Ausschlusskriterien:
- Schwelendes Multiples Myelom
- Aktive Plasmazellleukämie
- Amyloidose
- POEMS-Syndrom
- Stammzelltransplantation innerhalb von 12 Wochen vor der Aufnahme in die Studie
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt
Talquetamab
Talquetamab in Kombination mit Daratumumab oder in Kombination mit Daratumumab und Pomalidomid im Vergleich zu Daratumumab in Kombination mit Pomalidomid und Dexamethason bei Teilnehmern mit rezidiviertem oder refraktärem Multiplem Myelom (MonumenTAL-3)
Organisatorische Daten:
| Prüfplancode: | CR109082, 64407564MMY3002 |
| ISRCTN: | |
| EudraCT: | 2021-000202-22 |
| Clinicaltrials.gov: | NCT05455320 |
| DRKS: | |
| Sponsor: | Janssen |
| Studienphase: | 3 |
| Status: | Rekrutierung geplant bis Febr 2029 |
Ziel:
Ziel der Studie ist der Vergleich der Wirksamkeit von unter die Haut (subkutan, s.c.) verabreichtem Talquetamab in Kombination mit Daratumumab s.c. und Pomalidomid (Tal-DP) bzw. Talquetamab s.c. in Kombination mit Daratumumab s.c. (Tal-D) gegenüber Daratumumab s.c. in Kombination mit Pomalidomid und Dexamethason (DPd) bei Teilnehmern mit rezidiviertem oder refraktärem Multiplem Myelom, die mindestens eine vorherige Therapielinie erhalten haben.
Hintergrund:
Talquetamab ist ein humanisierter monoklonaler Antikörper, der gegen zwei Ziele gerichtet (bispezifisch) ist: gegen humanes CD3, ein Oberflächenantigen auf T-Zellen, und gegen GPRC5D (G protein-coupled receptor family C group 5 member D), ein Antigen in Tumorzellen oder auf deren Oberfläche (tumor-assoziiertes Antigen). Bei Verabreichung bindet Talquetamab sowohl an CD3 auf T-Zellen als auch an GPRC5D, das auf bestimmten Tumorzellen wie z.B. dem Multiplen Myelom vermehrt vorkommt und eine Schlüsselrolle bei der Vermehrung von Tumorzellen spielt.
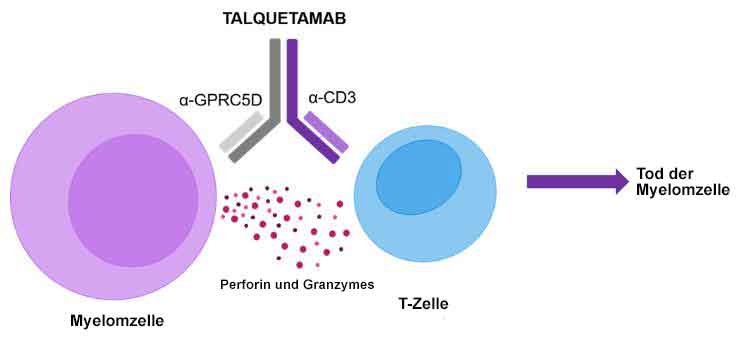
Abb. aus https://multiplemyelomahub.com/,
Durchführung:
Die Studie ist in drei Phasen unterteilt: Voruntersuchung (Screening), Behandlung (bis eines der folgenden Ereignisse eintritt: bestätigtes Fortschreiten der Krankheit, Tod, unverträgliche Toxizität, Widerruf der Einwilligung oder Ende der Studie) und Nachbeobachtung (bis eines der folgenden Ereignisse eintritt: Tod, Widerruf der Einwilligung, Verlust der Nachbeobachtung oder Ende der Studie). Wirksamkeit, Sicherheit, Pharmakokinetik, Immunogenität und Biomarker werden zu bestimmten Zeitpunkten untersucht. Die Gesamtdauer der Studie beträgt bis zu 6 Jahren und 6 Monaten.
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- Dokumentiertes Multiples Myelom, definiert als
- a) Diagnose des Multiplen Myeloms gemäß den diagnostischen Kriterien der International Myeloma Working Group (IMWG) und
- b) messbare Erkrankung zum Zeitpunkt des Screenings gemäß der Definition im Protokoll
- Rezidivierte oder refraktäre Erkrankung gemäß folgender Definition:
- eine rezidivierte Erkrankung ist definiert als anfängliches Ansprechen auf die vorherige Behandlung, gefolgt von einer nach IMWG-Kriterien bestätigten fortschreitenden (progredienten) Erkrankung mehr als 60 Tage nach Beendigung der Behandlung;
- eine refraktäre Erkrankung ist definiert als eine Verringerung des monoklonalen Paraproteins (M-Protein) um weniger als 25 % oder eine nach IMWG-Kriterien bestätigte fortschreitende Erkrankung während der vorherigen Behandlung oder ≤ 60 Tage nach Beendigung der Behandlung
- Teilnehmer müssen mindestens eine vorherige Linie einer Myelom-Therapie, einschließlich eines Proteasom-Inhibitors (PI) und Lenalidomid, erhalten haben. Patienten, die nur eine vorherige Linie einer Myelom-Therapie erhalten haben, müssen als Lenalidomid-refraktär gelten (d. h. bei ihnen ist bei Beendigung einer Lenalidomid-haltigen Therapie oder innerhalb von 60 Tagen danach eine fortschreitende (progrediente) Erkrankung nach IMWG-Kriterien nachweisbar). Teilnehmer, die 2 oder mehr vorherige Therapielinien zur Behandlung des Myeloms erhalten haben, müssen als Lenalidomid-behandelt gelten
- Dokumentierte Anzeichen einer fortschreitenden Erkrankung, basierend auf der Feststellung des Ansprechens durch den Prüfarzt gemäß den IMWG-Kriterien während oder nach der letzten Behandlung
- ECOG (Index zur Abstufung der Lebensqualität von Patienten mit Krebserkrankungen) von 0, 1 oder 2
Ausschlußkriterien
- Gegenanzeigen oder lebensbedrohliche Allergien, Überempfindlichkeiten oder Unverträglichkeiten gegenüber Hilfsstoffen des Studienmedikaments
- Refraktär gegenüber einem monoklonalen Antikörper gegen Cluster of Differentiation 38 (CD38), wie in den IMWG-Konsensrichtlinien definiert (Fortschreiten der Erkrankung während der Behandlung oder innerhalb von 60 Tagen nach Beendigung der Therapie mit einem monoklonalen Antikörper gegen CD38)
- Erhalt von Kortikosteroiden in einer maximalen Gesamtdosis von oder entsprechend ≥ 140 mg Prednison innerhalb von 14 Tagen vor Gabe der ersten Dosis des Studienmedikaments
- Bekannte aktive Beteiligung des zentralen Nervensystems (ZNS) oder klinische Anzeichen einer meningealen Beteiligung des Multiplen Myeloms. Bei Verdacht auf eine der beiden Erkrankungen sind eine negative Magnetresonanztomographie (MRT) des gesamten Gehirns und eine lumbale Zytologie erforderlich.
- Plasmazellleukämie zum Zeitpunkt der Voruntersuchung, Waldenström-Makroglobulinämie, Polyneuropathie, Organomegalie, Endokrinopathie, M-Protein und Hautveränderungen (POEMS-Syndrom) oder primäre Amyloid-Leichtketten-Amyloidose
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Charité - Universitätsmedizin Berlin der Freien Universität und der Humboldt-Universität, Campus Mitte
030-450-533222
Charité - Universitätsmedizin Berlin der Freien Universität und der Humboldt-Universität, Campus Virchow-Klinikum
PD Dr. Sturm, Fr. Stodder, 030-450-553862
Charite Campus Berlin-Buch
030 94892511
Studien
GMMG-HD8/DSMM-XIX
Induktionstherapie mit Lenalidomid, Bortezomib und Dexamethason und entweder intravenösem oder subkutanem Isatuximab bei Patienten mit neu diagnostiziertem Multiplem Myelom, die für eine Hochdosis-Chemotherapie mit nachfolgender autologer Stammzelltransplantation geeignet sind
Organisatorische Daten:
| Prüfplancode: | GMMG-HD 8 / DSMM XIX |
| ISRCTN: | |
| EudraCT: | 2022-000996-38 |
| Clinicaltrials.gov: | NCT05804032 |
| DRKS: | |
| Sponsor: | Universitätsklinikum Heidelberg DSMM, Sanofi und KKS-Netzwerk |
| Studienphase: | |
| Status: | Rekrutierung geplant bis Juni 2025 |
Ziel:
In dieser Studie für Patienten mit zuvor unbehandeltem Multiplem Myelom soll die Wirksamkeit der Induktionstherapie mit Isatuximab in Kombination mit Lenalidomid/Bortezomib/Dexamethason (RVd) bei Gabe von Isatuximab unter die Haut (subkutan) gegenüber einer Gabe in die Vene (intravenös) untersucht werden.
Durchführung:
Randomisierung: Die Patienten werden vor der Induktionstherapie nach dem Zufallsverfahren (randomisiert) einem von 2 Studienarmen (A oder B) zugeordnet.
- Patienten in Arm A erhalten 3 Zyklen des monoklonalen Antikörpers Isatuximab intravenös in Kombination mit dem RVd-Schema (Lenalidomid, Bortezomib, Dexamethason). Jeder Zyklus hat eine Dauer von 42 Tagen.
- Patienten in Arm B erhalten 3 Zyklen Isatuximab subkutan in Kombination mit RVd.
Nach der Induktionstherapie erhalten die Patienten eine Standard-Intensivierung (in der Regel eine Mobilisierungstherapie auf Cyclophosphamid-Basis, Stammzellenentnahme und hochdosiertes Melphalan, gefolgt von einer autologen Stammzelltransplantation
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- Bestätigte Diagnose eines neu diagnostizierten behandlungsbedürftigen Multiplen Myeloms gemäß den überarbeiteten Diagnosekriterien der International Myeloma Working Group (IMWG))
- Der Patient ist für eine Hochdosis-Chemotherapie und eine autologe Stammzelltransplantation geeignet
- Messbare Krankheitsaktivität (gemäß der Definition im Prüfplan)
- Alter 18 bis 70 Jahre bei Studieneinschluss
Ausschlußkriterien:
- Bekannte Überempfindlichkeit (oder Kontraindikation) gegen einen der Bestandteile der Studientherapie
- Systemische Amyloid-Leichtketten-Amyloidose (außer lokalisierte Amyloid-Leichtketten -Amyloidose, die auf die Haut oder das Knochenmark beschränkt ist)
- Plasmazell-Leukämie
- Vorherige Chemo- oder Strahlentherapie in den letzten 5 Jahren (außer lokaler Strahlentherapie bei lokalem Fortschreiten des Myeloms)
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Eine Phase-I-Studie mit MAGE-A1-spezifischen TCR-veränderten T-Zellen bei Patienten mit rezidiviertem/refraktärem multiplem Myelom
Organisatorische Daten:
| Prüfplancode: | MAGE-A1-TCR |
| ISRCTN: | |
| EudraCT: | 2017-001208-30 |
| Clinicaltrials.gov: | |
| DRKS: | DRKS00020221 |
| Sponsor: | Charité Campus Berlin Buch mit Förderung des Bundesministeriums für Bildung und Forschung |
| Studienphase: | Phase 1 |
| Status: | Rekrutierung läuft |
Ziel:
Untersuchung der Sicherheit und Verträglichkeit ansteigender Einzeldosen von genetisch veränderten, MAGE-A1-spezifischen, zytotoxischen autologen T-Zellen bei Patienten mit rezidiviertem und/oder refraktärem multiplem Myelom
Hintergrund:
T1367 T-Zellen sind autologe, zytotoxische (CD8+) T-Zellen, welche aus dem Blut entnommen und dann im Labor genetisch so verändert werden, dass sie gezielt Myelom-Zellen erkennen und zerstören können.
Durchführung
Die Studie besteht aus mehreren Phasen
1. Voruntersuchung
2. Vorlaufphase, in der T-Zellen mit einer Leukapherese aus dem Blut gefiltert und zu T1367 T-Zellen verändert werden (Dauer ca. 6 Wochen) sowie einer optionalen Überbrückungstherapie bei Patienten mit sehr aggressiver bzw. schnell fortschreitender Erkrankung,
3. Behandlungsphase mit Chemotherapie (Cyclophosphamid und Fludarabin) über 3 Tage und anschließender einmaliger intravenöser Verabreichung der T1367 T-Zellen sowie einer Nachuntersuchung. Während der Behandlungsphase sind die Patienten ca. 20 Tage stationär aufgenommen und werden engmaschig überwacht.
Es werden 3-6 Patienten pro Dosierungsstufe eingeschlossen. Jeder Patient wird nur in jeweils einer Dosisstufe behandelt.
Die vorgesehenen Dosierungsstufen sind 100.000, 1 Million, 10 Millionen und 50 bis 100 Millionen T-Zellen pro kg Körpergewicht (jeweils ± 20%).
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- Dokumentierte Diagnose eines rezidivierten/ refraktären multiplen Myeloms
- ECOG Performance Status von 0 bis 2 ((Index zur Abstufung der Lebensqualität von Patienten mit Krebserkrankungen)
- Mindestens 3 vorherige Therapielinien
- Nachgewiesene Expression von MAGE-A1 in mindestens 30% der Tumorzellen in einer Biopsie des Knochenmarks oder einer Manifestation außerhalb des Knochenmarks
Ausschlusskriterien:
- Allogene Stammzelltransplantation
- Jedwede Behandlung mit einer Gentherapie oder gentechnisch veränderten Immuntherapie
- Aktive oder chronische Hepatitis
- HIV Infektion
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Gemeinschaftspraxis Dr.med. Astrid Eilers-Lönnecker und Dr.med. Stephan Rackwitz
030-40999180
HELIOS Klinikum Berlin-Buch, Fachbereich Hämatologie und Stammzelltransplantation
030-9401-0
Studien
AlloRelapseMM
Allogene Stammzelltransplantation im Vergleich zur konventionellen Therapie als Salvage-Therapie für Patienten mit rezidiviertem / progredientem Multiplen Myelom nach einer Erstlinientherapie (AlloRelapseMMStudy)
Organisatorische Daten:
| Prüfplancode: | |
| ISRCTN: | |
| EudraCT: | 2021-001--5-67 |
| Clinicaltrials.gov: | NCT0567319 |
| DRKS: | |
| Sponsor: | Universitätsklinikum Hamburg-Eppendorf mit Unterstützung des Gemeinsamen Bundesausschusses (G-BA) |
| Studienphase: | III |
| Status: | Rekrutierung läuft, geplant bis 6/2027 |
Ziel:
In dieser Studie wird untersucht, ob die Transplantation von gespendeten Stammzellen (allogene Stammzelltransplantation) Vorteile gegenüber der gängigen Therapie bei Patienten mit Multiplem Myelom bietet, bei denen es nach einer Transplantation von körpereigenen Stammzellen (autologe Stammzelltransplantation) als Erstlinientherapie zu einem Erkrankungsrückfall (Rezidiv) oder einem Fortschreiten der Krankheit (Progression) gekommen ist. Es wird geprüft, ob die allogene Stammzelltransplantation das Gesamtüberleben bei diesen Patienten verlängert.
Hintergrund:
Eine Behandlungsoption des Multiplen Myeloms ist die Stammzelltransplantation. Nach einer Chemotherapie, bei der die Krebszellen zerstört werden, werden körpereigene (autologe) oder gespendete körperfremde (allogene) Stammzellen übertragen. Die übertragenen Stammzellen siedeln sich dann im Knochenmark an und bilden „frische“ Blutzellen. Die Stammzelltransplantation kommt als Behandlungsoption nach der ersten Diagnose (Erstlinientherapie), aber auch bei einem Rückfall nach Vorbehandlung (jenseits der Erstlinientherapie) in Frage. Auch wenn das Multiple Myelom oft nicht endgültig heilbar ist, soll die Therapie die Lebenserwartung verlängern und die Symptome zurückdrängen. (Quelle: https://www.g-ba.de/studien/erprobung/allorelapsemmstudy-studie/)
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- Diagnose Multiples Myelom,
- Rückfall (Rezidiv) oder Fortschreiten (Progress) des Multiplen Myeloms unter oder nach einer autologen Stammzelltransplantation
- Erhalt von höchstens 1 Zyklus einer sogenannten Salvage-Therapie (Rezidiv-Therapie) vor Einschluss in die Studie
Ausschlusskriterien:
- Unzureichende Organfunktion gemäß der Definition im Prüfplan
- Aktive Hepatitis B oder C oder unkontrollierte HIV-Infektion
- Andere aktive bösartige Erkrankung
- Vorherige Behandlung mit allogenen Stammzellen
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Zusätzliche Information
Myelomsprechstunde im Haupthaus B1, Erdgeschoss
Montag bis Freitag 7:30 bis 16:00 Uhr
Telefonische Voranmeldung (030) 94 01-52100 (Neuvorstellungen) -52120 (Ambulanz) erforderlich
MVZ Onkologischer Schwerpunkt am Oskar-Helene-Heim
030-644 99 52 30
Onkologisches Versorgungszentrum Friedrichshain, Dres. Lebahn, Maiwirth und Nawka
030- 80208770-0
Praxis am Volkspark Dr.med. Gunhild Kühn
Vivantes Klinik Spandau
030 130 10
Vivantes Klinikum Am Urban
030-130 21 0
Vivantes Klinikum Neukölln
030-130141466
Studien
GMMG-HD8/DSMM-XIX
Induktionstherapie mit Lenalidomid, Bortezomib und Dexamethason und entweder intravenösem oder subkutanem Isatuximab bei Patienten mit neu diagnostiziertem Multiplem Myelom, die für eine Hochdosis-Chemotherapie mit nachfolgender autologer Stammzelltransplantation geeignet sind
Organisatorische Daten:
| Prüfplancode: | GMMG-HD 8 / DSMM XIX |
| ISRCTN: | |
| EudraCT: | 2022-000996-38 |
| Clinicaltrials.gov: | NCT05804032 |
| DRKS: | |
| Sponsor: | Universitätsklinikum Heidelberg DSMM, Sanofi und KKS-Netzwerk |
| Studienphase: | |
| Status: | Rekrutierung geplant bis Juni 2025 |
Ziel:
In dieser Studie für Patienten mit zuvor unbehandeltem Multiplem Myelom soll die Wirksamkeit der Induktionstherapie mit Isatuximab in Kombination mit Lenalidomid/Bortezomib/Dexamethason (RVd) bei Gabe von Isatuximab unter die Haut (subkutan) gegenüber einer Gabe in die Vene (intravenös) untersucht werden.
Durchführung:
Randomisierung: Die Patienten werden vor der Induktionstherapie nach dem Zufallsverfahren (randomisiert) einem von 2 Studienarmen (A oder B) zugeordnet.
- Patienten in Arm A erhalten 3 Zyklen des monoklonalen Antikörpers Isatuximab intravenös in Kombination mit dem RVd-Schema (Lenalidomid, Bortezomib, Dexamethason). Jeder Zyklus hat eine Dauer von 42 Tagen.
- Patienten in Arm B erhalten 3 Zyklen Isatuximab subkutan in Kombination mit RVd.
Nach der Induktionstherapie erhalten die Patienten eine Standard-Intensivierung (in der Regel eine Mobilisierungstherapie auf Cyclophosphamid-Basis, Stammzellenentnahme und hochdosiertes Melphalan, gefolgt von einer autologen Stammzelltransplantation
Ein- und Ausschlusskriterien (Auswahl):
Einschlusskriterien:
- Bestätigte Diagnose eines neu diagnostizierten behandlungsbedürftigen Multiplen Myeloms gemäß den überarbeiteten Diagnosekriterien der International Myeloma Working Group (IMWG))
- Der Patient ist für eine Hochdosis-Chemotherapie und eine autologe Stammzelltransplantation geeignet
- Messbare Krankheitsaktivität (gemäß der Definition im Prüfplan)
- Alter 18 bis 70 Jahre bei Studieneinschluss
Ausschlußkriterien:
- Bekannte Überempfindlichkeit (oder Kontraindikation) gegen einen der Bestandteile der Studientherapie
- Systemische Amyloid-Leichtketten-Amyloidose (außer lokalisierte Amyloid-Leichtketten -Amyloidose, die auf die Haut oder das Knochenmark beschränkt ist)
- Plasmazell-Leukämie
- Vorherige Chemo- oder Strahlentherapie in den letzten 5 Jahren (außer lokaler Strahlentherapie bei lokalem Fortschreiten des Myeloms)
Alle weiteren Kriterien besprechen Sie bitte mit Ihrem behandelnden Arzt.
Unterkategorien
-
Berlin
- COM_STUDIES_NUM
- 12
